Transzkripciós faktorok¶
A sejtműködés, génexpresszió szabályozásában központi szerepet játszanak a transzkripciós faktorok (TF). Ezeknek a DNS-hez való kötődése központi mozzanat a transzkripció folyamatában. A TF-ek nukleinsavhoz való kapcsolódása meghatározott szekvenciákon (trancription factor binding site, TFBS) történhet meg. Azonban azt is tudjuk, hogy ugyanazon TF nem feltétlenül teljesen azonos TFBS-ekhez kapcsolódik a DNS különböző szakaszain. Ezeket a nem teljesen egységes bázissorrendű szekvenciákat motif-nak nevezik a szakirodalomban.
A MEF2 transzkripciós faktor esetén például az ismert kötőhelyek szekvenciája (Wasserman & Sandelin, 2004) az alábbi táblázatban látható:
| TFBS | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | G | A | C | C | A | A | A | T | A | A | G | G | C | A |
| 2 | G | A | C | C | A | A | A | T | A | A | G | G | C | A |
| 3 | T | G | A | C | T | A | T | A | A | A | A | G | G | A |
| 4 | T | G | A | C | T | A | T | A | A | A | A | G | G | A |
| 5 | T | G | C | C | A | A | A | A | G | T | G | G | T | C |
| 6 | C | A | A | C | T | A | T | C | T | T | G | G | G | C |
| 7 | C | A | A | C | T | A | T | C | T | T | G | G | G | C |
| 8 | C | T | C | C | T | T | A | C | A | T | G | G | G | C |
| i: | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
Ismert transzkripciós faktorok további lehetséges kötődési helyének meghatározásával (predikciójával) foglalkozó kutatásokban alkalmazott bioinformatikai elemzések ezekből az ismert TFBS-szekvenciákból indulnak ki. Kézenfekvő megközelítés lenne, hogy az ismert TFBS-szekvenciák konszenzusát használják a predikcióhoz.
library(Biostrings)
TFBSs.seqs = c('GACCAAATAAGGCA', 'GACCAAATAAGGCA', 'TGACTATAAAAGGA', 'TGACTATAAAAGGA',
'TGCCAAAAGTGGTC','CAACTATCTTGGGC', 'CAACTATCTTGGGC', 'CTCCTTACATGGGC')
TFBSs = DNAStringSet(TFBSs.seqs)
consensusString(TFBSs)
A konszenzus-szekvencia a nyolc kiindulási szekvencia információtartalmát ugyan sűrítette egyetlen IUPAC-kódolású szekvenciába, de egyúttal jelentősen csökkentette is az általa tárolt információt. Így növelve a kötési hely predikciójának megbízhatóságát. Pontosabb modelt jelent, ha a szekvencia minden pozíciójára vonatkozó nukleotidgyakorisági mátrixot (position frequency matrix, PFM) használunk:
consensusMatrix(TFBSs, baseOnly=TRUE)
| A | 0 | 4 | 4 | 0 | 3 | 7 | 4 | 3 | 5 | 4 | 2 | 0 | 0 | 4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | 3 | 0 | 4 | 8 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 2 | 4 |
| G | 2 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 6 | 8 | 5 | 0 |
| T | 3 | 1 | 0 | 0 | 5 | 1 | 4 | 2 | 2 | 4 | 0 | 0 | 1 | 0 |
| other | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
A consensusMatrix()-függvény által létrehozott PFM első négy sora tartalmazza az A, C, G, T bázisokra vonatkozó gyakorisági értékeket.
pfm.count = consensusMatrix(TFBSs, baseOnly=T)[1:4,]
A TFBSTools-csomag (Tan & Lenhard, 2016) tartalmaz speciális objek tumokat és függvényeket, amelyek segítségével könnyen dolgozhatunk a TFBS-elemzésekben.
library(TFBSTools)
pfm = PFMatrix(name='MEF2', profileMatrix=pfm.count)
pfm
An object of class PFMatrix
ID: Unknown
Name: MEF2
Matrix Class: Unknown
strand: +
Tags:
list()
Background:
A C G T
0.25 0.25 0.25 0.25
Matrix:
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12] [,13] [,14]
A 0 4 4 0 3 7 4 3 5 4 2 0 0 4
C 3 0 4 8 0 0 0 3 0 0 0 0 2 4
G 2 3 0 0 0 0 0 0 1 0 6 8 5 0
T 3 1 0 0 5 1 4 2 2 4 0 0 1 0
Vannak esetek, amikor a PFM-et nem gyakorisági értékekkel használják, hanem az egyes pozíciókban előforduló nukleotidok valószínűségével:
\(p(b,i) = \cfrac{f_{b,i}+s(b)}{N+ \sum_{b'\in\{A,C,G,T\}}s(b')}\ ,\)
ahol \(f_{b,i}\) a \(b\) nukleotid gyakorisága az \(i\) pozícióban; \(N\) a szekvenciák száma; \(p(b,i)\) a \(b\) nukleotid \(i\) pozícióban való előfordulásának korrigált valószínűsége; \(s(b)\) ún. pseudocount-függvény. Az átszámítást az alábbiak szerint végezhetjük:
pseudocount = sqrt(length(TFBSs.seqs))
pfm.prob = toPWM(pfm, type='prob', pseudocounts=pseudocount)
pfm.prob
An object of class PWMatrix
ID: Unknown
Name: MEF2
Matrix Class: Unknown
strand: +
Pseudocounts: 2.828427
Tags:
list()
Background:
A C G T
0.25 0.25 0.25 0.25
Matrix:
[,1] [,2] [,3] [,4] [,5] [,6] [,7]
A 0.06530097 0.43469903 0.43469903 0.06530097 0.34234952 0.71174758 0.43469903
C 0.34234952 0.06530097 0.43469903 0.80409709 0.06530097 0.06530097 0.06530097
G 0.25000000 0.34234952 0.06530097 0.06530097 0.06530097 0.06530097 0.06530097
T 0.34234952 0.15765048 0.06530097 0.06530097 0.52704855 0.15765048 0.43469903
[,8] [,9] [,10] [,11] [,12] [,13] [,14]
A 0.34234952 0.52704855 0.43469903 0.25000000 0.06530097 0.06530097 0.43469903
C 0.34234952 0.06530097 0.06530097 0.06530097 0.06530097 0.25000000 0.43469903
G 0.06530097 0.15765048 0.06530097 0.61939806 0.80409709 0.52704855 0.06530097
T 0.25000000 0.25000000 0.43469903 0.06530097 0.06530097 0.15765048 0.06530097
PFMatrix típusú objektumból egy
PWMatrix-objektum jött létre a toPWM()-fügvénnyel, aminek a
pseudocounts argumentumának Wasserman & Sandelin (2004)
ajánlásának megfelelően a TFBS szekvenciák számának négyzetgyökét
adtuk meg. A pseudocount értéke ettől eltérő is lehet.PWMatrix-nak nevezik, ez valójában még egy PFM. A PWM
adott nukleotid, adott pozícióbeli megfigyelt és a várható
háttér-valószínűségének \(log_2\) transzformált hányadosa:$W_{b,i} = :raw-latex:`\log`_2:raw-latex:cfrac{p(b,i)}{p(b)} , $
ahol \(p(b)\) a \(b\) nukleotid előfordulásának
háttér-valószínűsége; \(p(b,i)\) a \(b\) nukleotid \(i\)
pozícióbeli előfordulásának korrigált valószínűsége; \(W_{b,i}\) a
\(b\) nukleotid \(i\) pozícióbeli PWM-értéke. Az előző
toPWM()-függvény type='log2probratio' beállításával hozhatjuk
létre:
pwm = toPWM(pfm, type='log2probratio', pseudocounts=pseudocount)
pwm
An object of class PWMatrix
ID: Unknown
Name: MEF2
Matrix Class: Unknown
strand: +
Pseudocounts: 2.828427
Tags:
list()
Background:
A C G T
0.25 0.25 0.25 0.25
Matrix:
[,1] [,2] [,3] [,4] [,5] [,6] [,7]
A -1.9367518 0.7980888 0.7980888 -1.936752 0.4535419 1.5094376 0.7980888
C 0.4535419 -1.9367518 0.7980888 1.685442 -1.9367518 -1.9367518 -1.9367518
G 0.0000000 0.4535419 -1.9367518 -1.936752 -1.9367518 -1.9367518 -1.9367518
T 0.4535419 -0.6651985 -1.9367518 -1.936752 1.0760078 -0.6651985 0.7980888
[,8] [,9] [,10] [,11] [,12] [,13] [,14]
A 0.4535419 1.0760078 0.7980888 0.000000 -1.936752 -1.9367518 0.7980888
C 0.4535419 -1.9367518 -1.9367518 -1.936752 -1.936752 0.0000000 0.7980888
G -1.9367518 -0.6651985 -1.9367518 1.308939 1.685442 1.0760078 -1.9367518
T 0.0000000 0.0000000 0.7980888 -1.936752 -1.936752 -0.6651985 -1.9367518
A PWM alapján, adott szekvenciára kiszámolható egy összestett pontszám (site score), ami nagy számú és reprezentatív TFBS szekvencia esetén arányos a kötődési energiával (King & Roth, 2003).
\(S = \sum_{i=1}^w W_{l_i,i}\ ,\)
ahol \(S\) a szekvencia PWM-pontszáma; \(l_i\) az \(i\)
pozícióban lévő nukleotid; \(w\) a PWM szélessége. Például a fenti
PWM alapján a TTACATAAGTAGTC szekvenciára számított pontszám:
m = Matrix(pwm)
iseq = 'TTACATAAGTAGTC'
site.score = c()
for(i in 1:nchar(iseq)){
s = substr(iseq, i, i)
site.score = c(site.score, m[s, i])
}
site.score
- T
- 0.453541876358618
- T
- -0.665198492276212
- A
- 0.798088785639378
- C
- 1.68544162076245
- A
- 0.453541876358618
- T
- -0.665198492276212
- A
- 0.798088785639378
- A
- 0.453541876358618
- G
- -0.665198492276212
- T
- 0.798088785639378
- A
- 0
- G
- 1.68544162076245
- T
- -0.665198492276212
- C
- 0.798088785639378
sum(site.score)
A szekvencia összesített pontszámának abszolút érteke nem sokat mond arról, hogy a többi, már ismert kötőhely szekvenciájához hogyan viszonyul. Ezért az a szokás, hogy a PWM alapján számított legkisebb és legnagyobb összesített szekvencia pontszám alapján létrehozott eloszláshoz hasonlítják a vizsgált szekvencia összpontszámát. A példában szereplő PWM esetén az 5.26-os érték azt jelenti, hogy az összes lehetséges szekvencia 78%-a kisebb összesített pontszámmal rendelkezik. A PWM további fontos jellemzője az információtartalom profil (information content profile, IC, Schneider et al. (1986)). Az IC-t bitekben fejezik ki és a DNS-szekvencia esetében az értéke 0 és 2 bit között változhat. Egy olyan i pozícióban, ahol mindegyik nukleotid egyforma valószínűséggel fordulhat elő az IC = 0, míg egy olyan \(i\) pozícióban, ahol egyetlen nukleotid fordulhat elő az \(IC = 2\) (Bembom, 2017).
\(IC_i = 2 + \sum_b p_{b,i}\log_2 p_{b,i}\ ,\)
ahol IC i az i pozíció információ tartalma; a pb,i a b nukleotid i pozícióbeli korrigált valószínűsége. Azok a pozíciók, amelyek erősen konzervatívak és így kis toleranciával rendelkeznek a helyettesítésre vonatkozóan, magas IC-értékűek, míg a nagy helyettesítési toleranciájú pozíciók alacsony IC-értékűek (Bembom, 2017). A PFMatrix-ból ICMatrixot hozhatunk létre a toICM()-függvénnyel (A pseudocounts argumentumnak itt azért adtunk 0 értéket, hogy a Wasserman & Sandelin (2004) által bemutatott ábrát rekonstruáljuk):
icm = toICM(pfm, pseudocounts=0)
icm
An object of class ICMatrix
ID: Unknown
Name: MEF2
Matrix Class: Unknown
strand: +
Pseudocounts: 0
Schneider correction: FALSE
Tags:
list()
Background:
A C G T
0.25 0.25 0.25 0.25
Matrix:
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9]
A 0.0000000 0.29718047 0.5 0 0.3920872 1.2743811 0.5 0.1645207 0.43825316
C 0.1645207 0.00000000 0.5 2 0.0000000 0.0000000 0.0 0.1645207 0.00000000
G 0.1096805 0.22288535 0.0 0 0.0000000 0.0000000 0.0 0.0000000 0.08765063
T 0.1645207 0.07429512 0.0 0 0.6534787 0.1820544 0.5 0.1096805 0.17530126
[,10] [,11] [,12] [,13] [,14]
A 0.5 0.2971805 0 0.00000000 0.5
C 0.0 0.0000000 0 0.17530126 0.5
G 0.0 0.8915414 2 0.43825316 0.0
T 0.5 0.0000000 0 0.08765063 0.0
A PWM-ek grafikus reprezentációjának általánosan használt eszköze az ún. sequence logo (Schneider & Stephens, 1990). A logók a szekvencia mindegyik pozíciójára a négy nukleotidot jelölő betűhalmot tartalmaz. Az oszlop magassága a pozíció IC-jével, míg az egyes betűk magassága a nukleotid adott pozícióbeli relatív gyakoriságával arányos. A seqLogo()-függvénnyel létrehozhatunk szekvencialogokat. Ha a függvény ic.scale argumentumának TRUE értéket adunk, akkor a nukleotidoknak megfelelő oszlopok magassága az IC-vel arányos lesz.
seqLogo(icm, ic.scale=T)
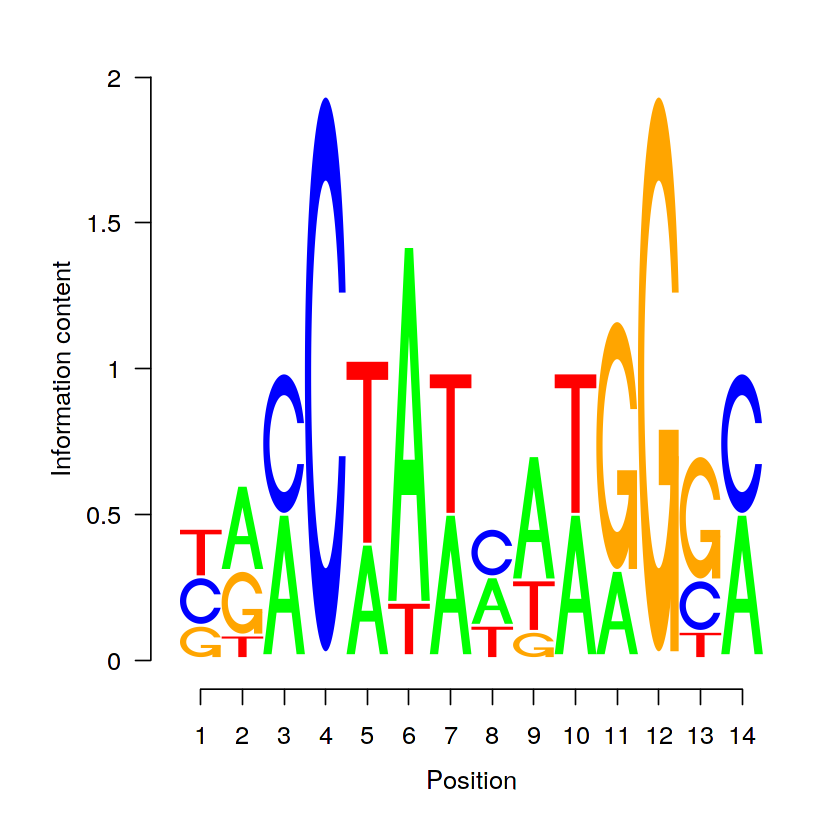
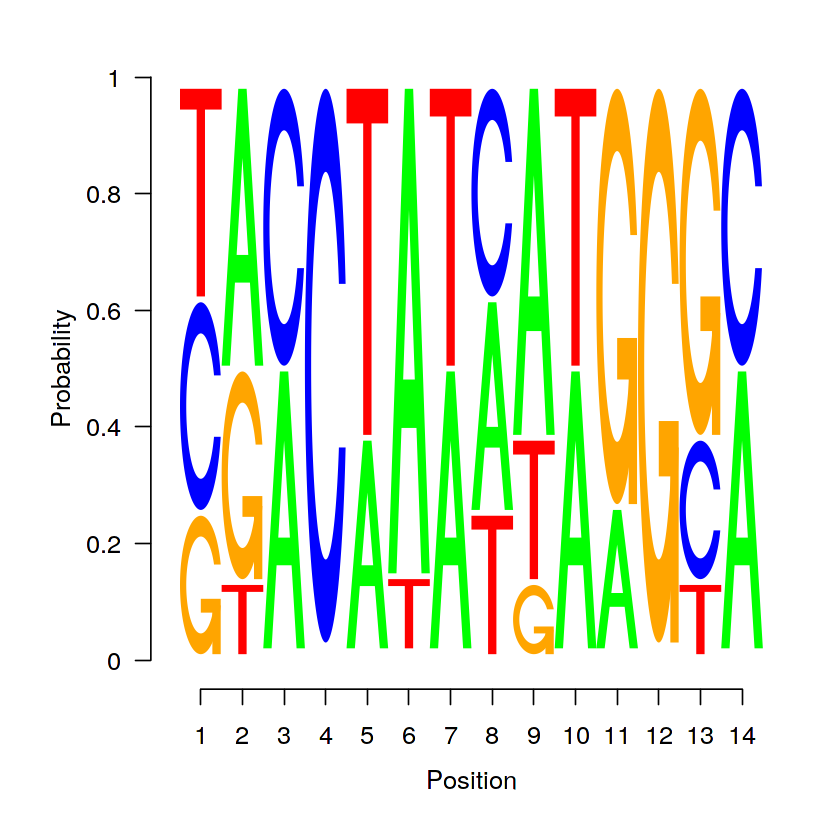
Ha ugyanennek az argumentumnak FALSE értéket adunk, akkor a logo oszlopai egyforma magasságúak lesznek és a betűk mérete a nukleotidok előfordulásának valószínűségét jelzi.
seqLogo(icm, ic.scale=F)
Motif-adatbázisok¶
MotifDb¶
Az ismert motif-ok jelenleg nem állnak rendelkezésre egyetlen szabad felhasználású forrásból, ezért gyűjtötték össze a különböző források, különböző liszenszelésű adatait a MotifDb-csomagba (Shannon, 2017). Az adatbázisból elérhető motifokra vonatkozó leíró inforációkat az alábbiak szerint kérdezhetjük le.
library(MotifDb)
MotifDb
See system.file("LICENSE", package="MotifDb") for use restrictions.
MotifDb object of length 8369
| Created from downloaded public sources: 2013-Aug-30
| 8369 position frequency matrices from 13 sources:
| cispb_1.02: 874
| FlyFactorSurvey: 614
| HOCOMOCOv10: 1066
| HOMER: 332
| hPDI: 437
| JASPAR_2014: 592
| JASPAR_CORE: 459
| jaspar2016: 1209
| jolma2013: 843
| ScerTF: 196
| stamlab: 683
| SwissRegulon: 684
| UniPROBE: 380
| 52 organism/s
| Hsapiens: 4094
| Mmusculus: 1251
| Dmelanogaster: 1147
| Scerevisiae: 876
| Athaliana: 351
| Celegans: 67
| other: 583
Scerevisiae-cispb_1.02-M0001_1.02
Scerevisiae-cispb_1.02-M0002_1.02
Scerevisiae-cispb_1.02-M0003_1.02
Csativa-cispb_1.02-M0004_1.02
Athaliana-cispb_1.02-M0005_1.02
...
Mmusculus-UniPROBE-Zfp740.UP00022
Mmusculus-UniPROBE-Zic1.UP00102
Mmusculus-UniPROBE-Zic2.UP00057
Mmusculus-UniPROBE-Zic3.UP00006
Mmusculus-UniPROBE-Zscan4.UP00026
Az adatbázisban elérhető mezők listája:
colnames(values(MotifDb))
- 'providerName'
- 'providerId'
- 'dataSource'
- 'geneSymbol'
- 'geneId'
- 'geneIdType'
- 'proteinId'
- 'proteinIdType'
- 'organism'
- 'sequenceCount'
- 'bindingSequence'
- 'bindingDomain'
- 'tfFamily'
- 'experimentType'
- 'pubmedID'
A motifok többféle eljárással kérdezhetők le az adatbázisból, a legegyszerűb a query()-függvény használata, pl. az összes humán motif kiolvasása:
query(MotifDb, 'Hsapiens')
MotifDb object of length 4094
| Created from downloaded public sources: 2013-Aug-30
| 4094 position frequency matrices from 10 sources:
| cispb_1.02: 313
| HOCOMOCOv10: 640
| hPDI: 437
| JASPAR_2014: 117
| JASPAR_CORE: 66
| jaspar2016: 442
| jolma2013: 710
| stamlab: 683
| SwissRegulon: 684
| UniPROBE: 2
| 1 organism/s
| Hsapiens: 4094
Hsapiens-jolma2013-BCL6B
Hsapiens-jolma2013-CTCF
Hsapiens-jolma2013-EGR1
Hsapiens-jolma2013-EGR1-2
Hsapiens-jolma2013-EGR2
...
Hsapiens-SwissRegulon-ZNF784.SwissRegulon
Hsapiens-SwissRegulon-ZNF8.SwissRegulon
Hsapiens-SwissRegulon-ZSCAN4.SwissRegulon
Hsapiens-UniPROBE-Sox4.UP00401
Hsapiens-UniPROBE-Oct_1.UP00399
Ha több szempotot is szeretnénk érvényesíteni a lekérdezésben, akkor egymásba ágyazott query()-ket használhatunk:
mot1 = query(query(query(MotifDb, 'Hsapiens'), 'JASPAR_2014'), 'MEF2')
mot1
MotifDb object of length 2
| Created from downloaded public sources: 2013-Aug-30
| 2 position frequency matrices from 1 source:
| JASPAR_2014: 2
| 1 organism/s
| Hsapiens: 2
Hsapiens-JASPAR_2014-MEF2C-MA0497.1
Hsapiens-JASPAR_2014-MEF2A-MA0052.2
A kiválasztott motifhoz tartozó mátrix(ok) az alábbi szerint olvasható(k) ki a mot1-objektumból:
as.list(mot1)
- $`Hsapiens-JASPAR_2014-MEF2C-MA0497.1`
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 A 0.3191489 0.33182435 0.19511091 0.1729289 0.0000000 0.73155274 0.77229516 0.953825260 0.9646899049 0.966500679 0.02535084 0.98551381 0.17609778 0.44137619 0.45676777 C 0.1453146 0.06835672 0.08872793 0.6392033 0.4459031 0.11588954 0.01448619 0.000000000 0.0000000000 0.000000000 0.02806700 0.00000000 0.05432322 0.37845179 0.20371209 G 0.3060208 0.25939339 0.37211408 0.0353101 0.0000000 0.03349932 0.10909914 0.039384337 0.0009053871 0.001810774 0.00000000 0.01448619 0.75645088 0.06699864 0.05703938 T 0.2295156 0.34042553 0.34404708 0.1525577 0.5540969 0.11905840 0.10411951 0.006790403 0.0344047080 0.031688547 0.94658216 0.00000000 0.01312811 0.11317338 0.28248076 - $`Hsapiens-JASPAR_2014-MEF2A-MA0052.2`
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 A 0.3265445 0.1147318 0.07875085 0.0000000 0.863543788 0.68431772 0.93890020 0.917854718 0.94365241 0.00407332 0.9993211134 0.15682281 0.3985064 0.35980991 0.2342159 C 0.1004752 0.1011541 0.80040733 0.3211134 0.046164291 0.00000000 0.00000000 0.003394433 0.00000000 0.00000000 0.0000000000 0.03326544 0.4290563 0.27766463 0.3048201 G 0.2572980 0.4243041 0.02172437 0.0000000 0.009504413 0.08078751 0.02647658 0.000000000 0.00000000 0.00000000 0.0000000000 0.78750849 0.0760353 0.07467753 0.1378140 T 0.3156823 0.3598099 0.09911745 0.6788866 0.080787508 0.23489477 0.03462322 0.078750849 0.05634759 0.99592668 0.0006788866 0.02240326 0.0964019 0.28784793 0.3231500
A mátrixokat az adatbázis PFM formában tárolja, mégpedig úgy, hogy a mátrix mindegyik oszlopához, vagyis a motif szekvenciájának pozíciójához tartozó értékeket egyre normálja. Amennyiben a mátrix létrehozásához felhasznált szekvenciák száma ismert, akkor azt a sequenceCount mezőből olvashatjuk ki:
as.matrix(values(mot1))
| providerName | providerId | dataSource | geneSymbol | geneId | geneIdType | proteinId | proteinIdType | organism | sequenceCount | bindingSequence | bindingDomain | tfFamily | experimentType | pubmedID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MEF2C | MA0497.1 | JASPAR_2014 | MEF2C | 4208 | ENTREZ | Q06413 | UNIPROT | Hsapiens | 2209 | NA | Other Alpha-Helix | MADS | ChIP-seq | 7559475 |
| MEF2A | MA0052.2 | JASPAR_2014 | MEF2A | 4205 | ENTREZ | Q02078 | UNIPROT | Hsapiens | 1473 | NA | Other Alpha-Helix | MADS | ChIP-seq | 1748287 |
Ennek felhasználásával előállítható pl. az első eredeti gyakorisági mátrix (PFM):
pfm1 = round(as.numeric(values(mot1)$sequenceCount[1])*as.list(mot1)[[1]])
pfm1
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 705 | 733 | 431 | 382 | 0 | 1616 | 1706 | 2107 | 2131 | 2135 | 56 | 2177 | 389 | 975 | 1009 |
| C | 321 | 151 | 196 | 1412 | 985 | 256 | 32 | 0 | 0 | 0 | 62 | 0 | 120 | 836 | 450 |
| G | 676 | 573 | 822 | 78 | 0 | 74 | 241 | 87 | 2 | 4 | 0 | 32 | 1671 | 148 | 126 |
| T | 507 | 752 | 760 | 337 | 1224 | 263 | 230 | 15 | 76 | 70 | 2091 | 0 | 29 | 250 | 624 |
JASPAR2014, JASPAR2016¶
A TFBSTools-könyvtár objektumaival való munkához létrehoztak JASPAR3 adatbázisokat (Tan, 2014, 2015). A JASPAR 2016 a 2014-hez képest több TF-kötő profilt tartalmaz, a korábbi adatokat frissítették benne, illetve 130 TFFM is van már benne (Mathelier et al., 2016). A 2014-es adatbázisból az összes motif lekérdezése:
library(JASPAR2014)
opts = list()
getMatrixSet(JASPAR2014, opts)
PFMatrixList of length 593
names(593): MA0004.1 MA0006.1 MA0008.1 MA0009.1 ... MA0599.1 MA0600.1 MA0113.2
A 2016-os adatbázisban elérhető motifok:
library(JASPAR2016)
getMatrixSet(JASPAR2016, opts)
PFMatrixList of length 1082
names(1082): MA0004.1 MA0006.1 MA0010.1 MA0011.1 ... MA1096.1 MA1097.1 MA1098.1
A TFBSTools-könyvtár getMatrixSet()-függvény második paraméterével állíthatjuk be a lekérdezési szempontokat, így a korábban használt motif-ot az alábbi szerint olvashatjuk ki:
opts = list(species=9606, name='MEF2A', all_versions=TRUE)
qres = getMatrixSet(JASPAR2016, opts)
qres
PFMatrixList of length 3
names(3): MA0052.1 MA0052.2 MA0052.3
Mivel a lekérdezés eredménye egy lista, ezért annak elemeit így írathatjuk ki:
qres[[1]]
An object of class PFMatrix
ID: MA0052.1
Name: MEF2A
Matrix Class: Other Alpha-Helix
strand: +
Tags:
$comment
[1] "-"
$family
[1] "MADS"
$medline
[1] "1748287"
$pazar_tf_id
[1] "TF0000034"
$tax_group
[1] "vertebrates"
$tfe_id
[1] "145"
$type
[1] "SELEX"
$collection
[1] "CORE"
$species
9606
"Homo sapiens"
$acc
[1] "EAX02249"
Background:
A C G T
0.25 0.25 0.25 0.25
Matrix:
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
A 1 0 57 2 9 6 37 2 56 6
C 50 0 1 1 0 0 0 0 0 0
G 0 0 0 0 0 0 0 0 2 50
T 7 58 0 55 49 52 21 56 0 2
A JASPAR2014-csomagban van egy JASPAR2014SitesSeqs-objektum is, aminek a segítségével egyszerűen lekérdezhető a motifok kiindulási szekvenciái:
JASPAR2014SitesSeqs$MA0052.1
A DNAStringSet instance of length 58
width seq names
[1] 26 ATGTGGGCTATTTATAGAAATTTCAG MA0052 MEF2A 1
[2] 26 GGAGTCGCCTCTTAACTATTTATAGA MA0052 MEF2A 2
[3] 26 AATGCGCGATGCACTATTTATAGTTC MA0052 MEF2A 3
[4] 27 CTATTTATAGCTAGGACGAGTCGTTCC MA0052 MEF2A 4
[5] 26 ACGCTTATTAAGTCTATTTATAGCCT MA0052 MEF2A 5
... ... ...
[54] 26 GAAATTCTGATTTATATTTAGACTCC MA0052 MEF2A 54
[55] 26 ATTTAACCCGAGTTACTTATAACTGC MA0052 MEF2A 55
[56] 25 GTTGGACGTGATGCTATTTTAGACA MA0052 MEF2A 56
[57] 26 TGTTACTATTTTAGTCCGAGTACTGT MA0052 MEF2A 57
[58] 26 ATCGGAAGGAAGTTGATCTATTTATA MA0052 MEF2A 58
Motifok, szekvenciák összehasonlítása¶
PFM-eket összehasonlíthatjuk a TFBSTools-könyvtár PFMSimilarity()-függvényével (Sandelin et al., 2003):
pfmORIG = qres[[1]]
PFMSimilarity(pfmORIG, pfmORIG)
- score
- 20
- relScore
- 100
A score itt látható 100%-os egyezés (relScore) esetén a szekvencia hosszának kétszerese. Ha egy apró módosítást végzünk az egyik mátrixon, akkor az alábbi eredményt kapjuk:
tmp = Matrix(pfmORIG)
tmp[2,2] = 1000
tmp[4,2] = 22
pfmALT = PFMatrix(profileMatrix = tmp)
PFMSimilarity(pfmORIG, pfmALT)
- score
- 18.0851783752441
- relScore
- 90.4258918762207
Ugyanezzel a függvénnyel összehasonlíthatunk egy PFM-et és egy IUPAC-szekvenciát is, pl. a PFM-et és annak konszenzus szekvenciáját:
library(seqLogo)
pwmORIG = toPWM(pfmORIG, type='prob')
pwmALT = toPWM(pfmALT, type='prob')
seq = makePWM(Matrix(pwmORIG))@consensus
seq
PFMSimilarity(pfmORIG, seq)
- score
- 19.5939350128174
- relScore
- 97.9696750640869
PWM-ek összehasonlítására a TFBSTools-könyvtár PWMSimilarity()-függvényével három különböző módszeret használhatunk. Normalizált euklideszi távolság:
PWMSimilarity(pwmORIG, pwmALT, method='Euclidean')
A statisztika értéke 0 és 1 között változhat, a 0 teljes egyezőséget, az 1 teljes eltérést jelent.
Pearson-korreláció:
PWMSimilarity(pwmORIG, pwmALT, method='Pearson')
A statisztika értéke -1 és 1 között változhat, az 1 teljes egyezőséget, a −1 teljes eltérést jelent.
Kullback-Leibler eltérés (Linhart et al., 2008):
PWMSimilarity(pwmORIG, pwmALT, method='KL')
A két PWM eloszlásának eltérését vizsgáljuk ezzel a mértékkel, értéke 0 − 1 között változhat, a 0 erős hasonlóságot, az 1 nagy eltérést jelez.
PWM keresése szekvenciában¶
A searchSeq()-függvénnyel vizsgálhatjuk, hogy a PWM-ünk mely DNS-szakaszra illeszkedik. Ha a fenti példa konszenzusszekvenciáját beillesztjük egy véletlen szekvenciába, az alábbi eredményt kapjuk:
set.seed(20)
rnd.seq.str = paste(sample(c('A', 'C', 'G', 'T'), 30, replace=TRUE), collapse='')
seq.str = paste(substr(rnd.seq.str, 1, 10), seq, substr(rnd.seq.str, 11, 30), sep='')
seq.str
seq.dns = DNAString(seq.str)
siteset = searchSeq(pwmORIG, seq.dns, seqname='teszt', strand='*', min.score=0.8)
siteset
An object of class SiteSet with 2 site sequences
seqname source feature start end score strand frame
1 teszt TFBS TFBS 11 20 8.877551 + .
2 teszt TFBS TFBS 11 20 7.414966 - .
attributes
1 TF=MEF2A;class=Other Alpha-Helix;sequence=CTATTTATAG
2 TF=MEF2A;class=Other Alpha-Helix;sequence=CTATAAATAG
A min.score-argumentumban meghatározhatjuk, hogy a PWM és a vizsgált szekvencia között milyen mértékű eltérés fogadható még el, 0 − 1 vagy 0 − 100% közötti érték lehet. A PWM-ből létrehozható szekvenciák minimális és maximális összesített pontszámából számított eloszlásnak az illeszkedő szakaszok szűrésére használt percentilis határértéket jelenti.
A függvény eredménye egy SiteSet-objektum, azonban, ha az első argumentuma nem PWMatrix, hanem PWMatrixList, akkor SiteSetList-objektumot kapunk vissza.
qres.pwm = toPWM(qres, type='prob')
siteset = searchSeq(qres.pwm, seq.dns, seqname='teszt', strand='*', min.score=0.8)
siteset
SiteSetList of length 3
names(3): MA0052.1 MA0052.2 MA0052.3
A PWM szekvenciával való összehasonlítására használhatjuk a Biostrings-csomag függvényeit is:
pwm.B = PWM(Matrix(pfmORIG))
pwm.B
| A | 0.03016684 | 0.0000000 | 0.10184813 | 0.04118415 | 0.06768201 | 0.06033369 | 0.09379254 | 0.04118415 | 0.10151784 | 0.06033369 |
|---|---|---|---|---|---|---|---|---|---|---|
| C | 0.09940363 | 0.0000000 | 0.03016684 | 0.03016684 | 0.00000000 | 0.00000000 | 0.00000000 | 0.00000000 | 0.00000000 | 0.00000000 |
| G | 0.00000000 | 0.0000000 | 0.00000000 | 0.00000000 | 0.00000000 | 0.00000000 | 0.00000000 | 0.00000000 | 0.04118415 | 0.09940363 |
| T | 0.06311563 | 0.1021727 | 0.00000000 | 0.10118162 | 0.09902686 | 0.10013519 | 0.08327179 | 0.10151784 | 0.00000000 | 0.04118415 |
res.B = matchPWM(pwm.B, seq.dns, with.score=TRUE, min.score=0.8)
res.B
Views on a 40-letter DNAString subject
subject: TTCGTTAACCCTATTTATAGGTAGACCACTCACACATTAG
views:
start end width
[1] 11 20 10 [CTATTTATAG]
A Biostrings-csomagra épülően a PWMEnrich-csomag (Stojnic & Diez, 2015) további lehetőségeket nyújt PWM-ek szekvenciákra való illeszkedésének vizsgálatában. A csomag függvényeivel többek között azt vizsgálhatjuk (a Clover-hez hasonlóan (https: //zlab.bu.edu/clover/), hogy egy adott szekcvenciában mely motif-ok vannak felülreprezentálva. Ahogy korábban láttuk a motif-oknak valamely szekvenciára való kötődését használhatjuk a TFBS predikciójára. Azonban a fent bemutatott módszer, illetve a legtöbb, a szakirodalomban fellelhető eljárás esetén meg kell határoznunk valamilyen a motif és a szekvencia hasonlóságára vonatkozó határértéket. Ahogy Frith et al. (2004) bemutatja, ha ez az érték alacsony, akkor sok nem „valódi” TF-et azonosíthatunk, mint olyat, ami a kérdéses szekvenciához kötődhet. Ha pedig túl magas, akkor „valóban” kötödő TF-eket veszíthetünk el. Azonkívűl ezek az eljárások nem kezelik azt, ha egy TF az adott szekvencián egynél több TFBS-el rendelkezik, illetve azt sem, hogy a teljes genomban vagy egy-egy kromoszómán milyen valószínűséggel kötődhet. Frith et al. (2004) által bemutatott megközelítésben a TF-ekre vonatkozóan egy nagyobb genomszakaszra, vagy a teljes genomra meghatározzuk, hogy milyen ún. nyers pontszámmal (raw score) kötődnek. Majd a vizsgált szekvenciára is meghatározzuk ezt a pontszámot. Így a háttér ismeretében becsülhető, hogy milyen valószínűséggel kötődhet a TF a vizsgált szekvencián olyan (vagy annál nagyobb) nyers pontszámmal, amilyet számoltunk rá. Frith et al. (2004) szerint a p < 0.01 valószínűséggel kötődő TF-ek esetén alaposan feltételezhető, hogy az adott szakaszon felülreprezentált (over-represented), míg ha a p > 0.99, akkor alulreprezentált (under-represented). Továbbá, ha ilyen felül- vagy alulreprezentáltságot tapasztalunk, akkor annak funkcionális szerepe lehet. Mondjuk, hogy egy a humán 8. kromoszómán lévő szekvenciára kötődő TF-eket szeretnénk azonosítani. Ráadásul nem az összes ismert TF-re vagyunk kiváncsiak, hanem csak néhányra. Először a TF-eket kell megfelelő formátumba alakítani:
library(TFBSTools)
library(JASPAR2016)
opts = list(
species=9606,
name=c('AR', 'ETV1', 'FOXA1', 'GATA2', 'HOXB13', 'NKX3-1'),
all_versions=TRUE
)
TF.set = getMatrixSet(JASPAR2016, opts)
lst = as.list(TF.set)
TF.lst = lapply(lst, Matrix)
names(TF.lst) = paste(as.character(lapply(lst, name)), names(TF.lst), sep='_')
A 8. humán kromoszómára lesz szükségünk a háttér létrehozásához, azonban mivel a szekvencia tartalmaz N-eket, ezeket ki kell törölnünk a továbbiakban használt függvényekhez. Ezt – többek között – tehetjük az alábbiak szerint:
library(BSgenome.Hsapiens.UCSC.hg19)
chr8 = as.character(BSgenome.Hsapiens.UCSC.hg19$chr8)
lst = strsplit(chr8, 'N')
lst = lst[[1]]
lst = lst[which(lapply(lst, nchar)!=0)]
chr8 = DNAStringSet(unlist(lst))
A PWMEnrich-könyvtár makePWMLognBackground()-függvényével hozhatunk létre saját lognormális háttér eloszlást valamely szekvenciá(k)ra, itt a 8. kromoszómára:
library(PWMEnrich)
bg.logn = makeBackground(motifs=TF.lst, bg.seq=chr8, type='logn', algorithm='human')
A motifok szekvencián belüli alul- vagy felülreprezentáltságát ezek után az alábbiak szerint vizsgálhatjuk:
myseq = DNAString('TGCGTTCTCTTGGAGACTCTCTATTTGCCCCTCTCTTTTACAG')
res = motifEnrichment(myseq, bg.logn)
rpt = sequenceReport(res, 1)
rpt
Scanning sequence 1 / 1
Calculating motif enrichment scores ...
An object of class 'MotifEnrichmentReport':
rank target id raw.score p.value
1 1 FOXA1_MA0148.1 FOXA1_MA0148.1 0.3001255927 0.3072201
2 2 AR_MA0007.2 AR_MA0007.2 0.0400636241 0.3281074
3 3 FOXA1_MA0148.2 FOXA1_MA0148.2 0.2398628307 0.3330050
4 4 FOXA1_MA0148.3 FOXA1_MA0148.3 0.0020333381 0.5656657
5 5 GATA2_MA0036.2 GATA2_MA0036.2 0.0002374392 0.6262785
6 6 NKX3-1_MA0124.1 NKX3-1_MA0124.1 0.0250463314 0.7531180
7 7 ETV1_MA0761.1 ETV1_MA0761.1 0.0002927674 0.9671194
8 8 HOXB13_MA0901.1 HOXB13_MA0901.1 0.0005775918 0.9709271
9 9 GATA2_MA0036.1 GATA2_MA0036.1 0.0426125957 0.9991309
Az eredményeket a plot()-függvénnyel vizualizálhatjuk is. A listából és az ábráról az látható, hogy egyedül a GATA2_MA0036.1 tekinthető alulreprezentáltnak.
plot(rpt)

„de novo” motif-azonosítás¶
Új motifok keresésére a TFBSTools-csomag is tartalmaz függvényt (runMEME), ami a MEME-t (http://meme-suite.org/) használja. Ehhez képest újabb és talán hatékonyabb módszert fejlsztett Li (2009), amit az rGADEM-csomag (Droit et al., 2014) segítségével alkalmazhatunk az R-en belül. A vizsgálatokból (pl. ChIP-seq) származó szekvencákat az alábbiak szerint elemezhetjük motif-tartalmukra vonatkozóan.
Az elemzésekhez szükségünk van a szekvenciákra, amelyeket az itt bemutatott példában FASTA-fájlban tárolunk, az állomány első három sora így néz ki:
>FOXA1 _ 1
CTACAGCTGTTCCTTGTCATCAGCCTGGGGGTGGGTAGTATTTTGATCTTAACGATGCCTGTTTGTTTACTCTGAGCTAGTCTTAGTGTAAGAGTCACCTC
TCTATGTACATAGAAACCGTTGTCCTTTATTGACAGAAGCCCTGGAGATGGGTCCCCATGTGACTGTAGGGTTCCTGAAACCTGGCAGGCCACTCTGCTTG
>FOXA1 _ 10
TTATTCTGATGTGGTTTTGCGGTTATACAGTAAGCAGCACTGCTTATGTGGACATGGTGTACTTTCAGTTTCTGAAAGTGAGTCATGTTGACTTTCCTCTG
AGGAGTAAGAGTGACCCTTGCTTAAAAGGACAACATGACTAGAAGGAAGAAACACCAGAGGCCACCAGGACCAGAATGTTTACCAATGTAGGCAGTCACTA
>FOXA1 _ 11
AAAGGAGAAACACAGCCAAATAATAAAACAATATCTTCTGTAAGTAAAGAGTACACCCCTGTTTACCTGGTCGCCACTGTTTATTCTGAAAGACTACACTA
AGCAAATACTGAGCCTGACAGCTAGGCTGGAGGGGAGGGGTCTCTAGGCCACAAAGGTGCAAAGCCCTCTTTCAGATCCATCTCCACCATTTCCCTTCAGG
Az R-ben így olvassunk be a szekvenciákat és alakítjuk át Biostrings objektummá:
library(rGADEM)
fasta.file = system.file('extdata/Test_100.fasta', package='rGADEM')
seqs = readDNAStringSet(fasta.file, 'fasta')
seqs
A DNAStringSet instance of length 49
width seq names
[1] 202 CTACAGCTGTTCCTTGTCATCAG...ACCTGGCAGGCCACTCTGCTTG FOXA1 _ 1
[2] 202 TTATTCTGATGTGGTTTTGCGGT...TTACCAATGTAGGCAGTCACTA FOXA1 _ 10
[3] 202 AAAGGAGAAACACAGCCAAATAA...ATCTCCACCATTTCCCTTCAGG FOXA1 _ 11
[4] 202 TGTACCCCCCCAATATTTCATGA...AACACTGAGCCTGGCATTCCAA FOXA1 _ 12
[5] 202 TTTAAGACTGCCACCTGAAATCA...AGAAGACGGGTTGAGCGAGTCA FOXA1 _ 13
... ... ...
[45] 202 ATTAGTTCATGCCAGGCAGGGTT...GTGGTTTGGAAACACCCTCATG FOXA1 _ 5
[46] 202 CCAGAGCCACCACAGCCAGGCCT...CCTGAAAACACCTGCTGTTTAA FOXA1 _ 6
[47] 202 CAGATCAGAGCCTGGGAGCGGGC...AGGGAAGGGAGCTGTGGGAGGA FOXA1 _ 7
[48] 202 GGAATGTGATTTACCCAGATAAA...AAACATGTCCACATGGAACCTG FOXA1 _ 8
[49] 202 GGCTTTTCCAAGAACAATAGTGT...TAAAGTCGGGTTGGTCCTTGGC FOXA1 _ 9
Továbbá szükség van a referenciaként szolgáló genomra:
library(BSgenome.Hsapiens.UCSC.hg19)
Hsapiens
Human genome:
# organism: Homo sapiens (Human)
# provider: UCSC
# provider version: hg19
# release date: Feb. 2009
# release name: Genome Reference Consortium GRCh37
# 93 sequences:
# chr1 chr2 chr3
# chr4 chr5 chr6
# chr7 chr8 chr9
# chr10 chr11 chr12
# chr13 chr14 chr15
# ... ... ...
# chrUn_gl000235 chrUn_gl000236 chrUn_gl000237
# chrUn_gl000238 chrUn_gl000239 chrUn_gl000240
# chrUn_gl000241 chrUn_gl000242 chrUn_gl000243
# chrUn_gl000244 chrUn_gl000245 chrUn_gl000246
# chrUn_gl000247 chrUn_gl000248 chrUn_gl000249
# (use 'seqnames()' to see all the sequence names, use the '$' or '[[' operator
# to access a given sequence)
A két forrásállományt a GADEM-függvénnyel elemezhetjük:
gadem.res = GADEM(seqs, genome=Hsapiens)
top 3 4, 5-mers: 10 28 52
top 3 4, 5-mers: 8 24 44
gadem.res
Object of class 'gadem'
This object has the following slots:
motifs,pwm,consensus,align,name,seq,chr,start,end,strand,seqID,pos,pval,fastaHeader
A számítások eredményeként az „új motifra” vonatkozóan létrejött objektumból többek között kiolvasható a PWM:
getPWM(gadem.res)
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0.4679 | 0.3542 | 0.4427 | 0.0005 | 0.0005 | 0.7206 | 0.9985 | 0.9859 | 0.0005 | 0.9985 | 0.2532 |
| C | 0.0510 | 0.1900 | 0.0131 | 0.0005 | 0.3416 | 0.2784 | 0.0005 | 0.0005 | 0.7458 | 0.0005 | 0.2658 |
| G | 0.0131 | 0.3416 | 0.0384 | 0.9985 | 0.0005 | 0.0005 | 0.0005 | 0.0131 | 0.0005 | 0.0005 | 0.2532 |
| T | 0.4679 | 0.1142 | 0.5058 | 0.0005 | 0.6574 | 0.0005 | 0.0005 | 0.0005 | 0.2532 | 0.0005 | 0.2279 |
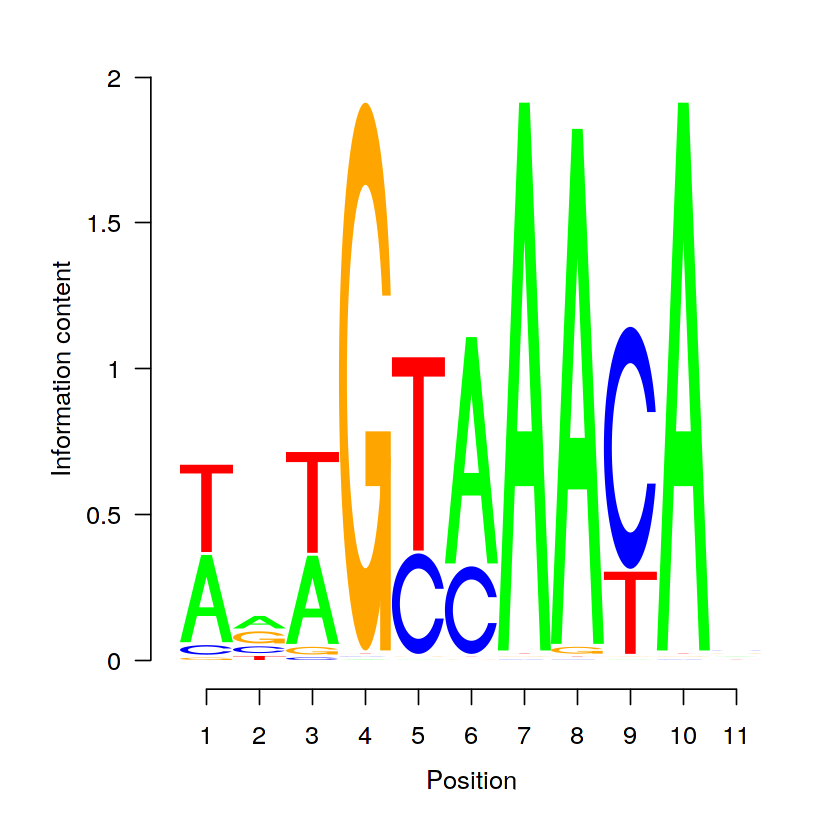
A gadem.res-objektumból közvetlenül létrehozhatunk szekvencialogot az rGADEM-könyvtár saját plot()-függvényével, azonban ez nem túl rugalmas, így az ábrázolásban inkább érdemes a seqLogo-könyvtár függvényét használni:
pwm.m = getPWM(gadem.res)[[1]]
seqLogo::seqLogo(pwm.m)
motifStack¶
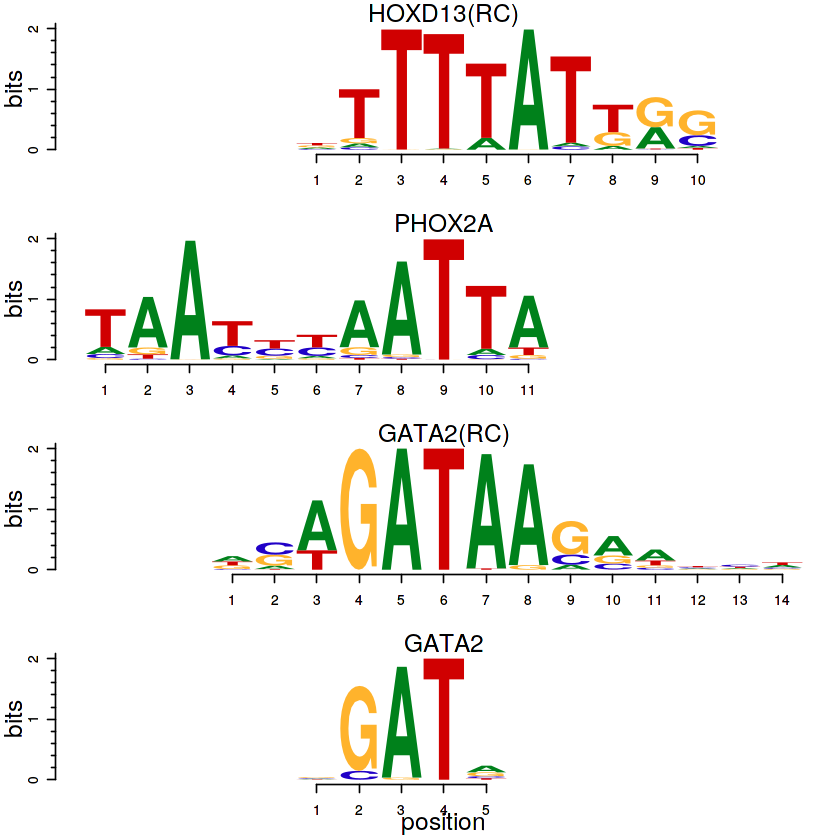
Több motif együttes ábrázolására hozták létre a motifStack-csomagot (Ou & Zhu, 2017). A csomag függvényei nagy rugalmasságot nyújtanak a vizualizációra, ezek közül egy egyszerűbb példa azt mutatja be, hogy egymással valamilyen szintű hasonlóságot mutató motifokat hogyan tudjuk megjeleníteni:
opts = list(species=9606, name=c('GATA2', 'PHOX2A', 'HOXD13'), all_versions=T)
qres = getMatrixSet(JASPAR2016, opts)
library(motifStack)
pfm.lst = list()
for(i in length(qres):1){
mo = qres[[i]]
m = Matrix(mo)
pfm.lst[[i]] = new("pfm", mat=pcm2pfm(m), name=name(mo))
}
pfm.lst = DNAmotifAlignment(pfm.lst, threshold=0.8)
plotMotifLogoStack(pfm.lst)
TFBS-en belüli SNP-k hatásának predikciója¶
Ahogy a genomban általánosságban, úgy a TF-kötőhelyeken is bekövetkeznek szekvenciális változások. Ennek ismeretében könnyen merül fel az a kérdés, hogy ezek a változások hogyan befolyásolják a génexpressziót? A kérdés megválaszolásához kapcsolódó motifbreakR-csomag (Coetzee et al., 2015) segítséget nyújt annak eldöntésében, hogy a polimorfizmus/mutációkörüli szekvencia jó kötést biztosít-e, illetve, hogy a megváltozott allél információvesztéssel, vagy -nyereséggel jár-e.
Polimorfizmus adatok¶
Szekvencia polimorfizmusokra, variánsokra vonatkozóan számos adatbázis áll rendelkezésre. Ilyen pl. az NCBI dbSNP-je, amelyből a Bioconductor-on elérhetünk számos verziót:
library(BSgenome)
available.SNPs()
- 'SNPlocs.Hsapiens.dbSNP.20101109'
- 'SNPlocs.Hsapiens.dbSNP.20120608'
- 'SNPlocs.Hsapiens.dbSNP141.GRCh38'
- 'SNPlocs.Hsapiens.dbSNP142.GRCh37'
- 'SNPlocs.Hsapiens.dbSNP144.GRCh37'
- 'SNPlocs.Hsapiens.dbSNP144.GRCh38'
- 'SNPlocs.Hsapiens.dbSNP149.GRCh38'
- 'SNPlocs.Hsapiens.dbSNP150.GRCh38'
- 'XtraSNPlocs.Hsapiens.dbSNP141.GRCh38'
- 'XtraSNPlocs.Hsapiens.dbSNP144.GRCh37'
- 'XtraSNPlocs.Hsapiens.dbSNP144.GRCh38'
Míg az SNPlocs-állományok csak SNP-ket (single nucleotide-polymorphism) tartalmaznak, addig az XtraSNPlocs-állományokban további molekuláris változatok (pl. in-del, multinucleotide-polymorphism) találhatók. Az SNPlocs-állományokból több módon lekérdezhetők SNP-k, pl. azonosító alapján:
library(SNPlocs.Hsapiens.dbSNP150.GRCh38)
snpsById(SNPlocs.Hsapiens.dbSNP150.GRCh38, 'rs7837328', ifnotfound='drop')
GPos object with 1 position and 2 metadata columns:
seqnames pos strand | RefSNP_id alleles_as_ambig
<Rle> <integer> <Rle> | <character> <character>
[1] 8 127410882 * | rs7837328 R
-------
seqinfo: 25 sequences (1 circular) from GRCh38.p7 genome
Az rs7837328 azonosítójú SNP a lekérdezés szerint a 8. kromoszómán helyezkedik el, és vagy A vagy G lehet. Az SNP-azonosítót használhatjuk az rs előtag nélkül is, akár karakterként, akár egész számként. Lekérdezhetjük azt is, hogy egy adott szekvenciaszakasz tartalmaz-e ismert SNP-t. Ehhez először létre kell hoznunk egy GRanges-objektumot amit kereshetünk a SNP-adatbázisban:
gr = GRanges(
seqnames=c('8'),
ranges=IRanges(127410880, 127410885),
strand=c('*')
)
snpsByOverlaps(SNPlocs.Hsapiens.dbSNP150.GRCh38, ranges=gr)
GPos object with 1 position and 2 metadata columns:
seqnames pos strand | RefSNP_id alleles_as_ambig
<Rle> <integer> <Rle> | <character> <character>
[1] 8 127410882 * | rs7837328 R
-------
seqinfo: 25 sequences (1 circular) from GRCh38.p7 genome
Az XtraSNPlocs-állományokból hasonlóan olvashatunk ki adatokat. A 8. kromoszómán azonosított variánsok pl.:
q = snpsBySeqname(
XtraSNPlocs.Hsapiens.dbSNP144.GRCh38,
'ch8',
columns=c('RefSNP_id', 'snpClass')
)
q
GRanges object with 606507 ranges and 2 metadata columns:
seqnames ranges strand | RefSNP_id snpClass
<Rle> <IRanges> <Rle> | <character> <factor>
[1] ch8 [61481, 61482] + | rs200678772 in-del
[2] ch8 [61793, 61793] + | rs752636065 in-del
[3] ch8 [70901, 70901] + | rs201602045 in-del
[4] ch8 [70950, 70950] + | rs767986677 in-del
[5] ch8 [73088, 73088] + | rs750775551 in-del
... ... ... ... . ... ...
[606503] ch8 [145076006, 145076005] + | rs145524841 in-del
[606504] ch8 [145076464, 145076463] + | rs199775790 in-del
[606505] ch8 [145077216, 145077215] + | rs200565575 in-del
[606506] ch8 [145078174, 145078173] + | rs574365224 in-del
[606507] ch8 [145078175, 145078174] + | rs539896939 in-del
-------
seqinfo: 25 sequences (1 circular) from GRCh38.p2 genome
A variánsok típusainak megoszlása a 8. kromoszómán:
table(elementMetadata(q)$snpClass)
in-del heterozygous
596293 0
microsatellite named-locus
215 0
no-variation mixed
0 0
multinucleotide-polymorphism
9999
Irodalomjegyzék¶
Bembom, O. (2017). seqLogo: Sequence logos for DNA sequence alignments. R package version 1.42.0.
Coetzee, S. G., Coetzee, G. A., & Hazelett, D. J. (2015). motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics, 31(23), 3847–3849.
Droit, A., Gottardo, R., Robertson, G., & Li, L. (2014). rGADEM: de novo motif discovery. R package version 2.24.0.
Frith, M. C., Fu, Y., Yu, L., Chen, J., Hansen, U., & Weng, Z. (2004). Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Research, 32(4), 1372–1381.
King, O. D. & Roth, F. P. (2003). A non-parametric model for transcription factor binding sites. Nucleic Acids Res, 31, e116.
Li, L. (2009). GADEM: A Genetic Algorithm Guided Formation of Spaced Dyads Coupled with an EM Algorithm for Motif Discovery. Journal of Computational Biology, 16(2), 317–329.
Linhart, C., Halperin, Y., & Shamir, R. (2008). Transcription factor and microRNA motif discovery: the Amadeus platform and a compendium of metazoan target sets. Genome research, 18(7), 1180–1189.
Mahony, S., Auron, P. E., & Benos, P. V. (2007). DNA familial binding profiles made easy: comparison of various motif alignment and clustering strategies. PLoS Computational Biology, 3(3), e61.
Mahony, S. & Benos, P. V. (2007). STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acid Research, 35, W253–258.
Mathelier, A., Fornes, O., Arenillas, D. J., Chen, C., Denay, G., Lee, J., Shi, W., Shyr, C., Tan, G., Worsley-Hunt, R., Zhang, A. W., Parcy, F., Lenhard, B., Sandelin, A., & Wasserman, W. W. (2016). JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Research, 44(Database-Issue), 110–115.
Mathelier, A. & Wasserman, W. W. (2013). The next generation of transcription factor binding site prediction. PLOS Computational Biology, 9(9), 1–18.
Mercier, E. & Gottardo, R. (2014). MotIV: Motif Identification and Validation. R package version 1.32.0.
Ou, J. & Zhu, L. J. (2017). motifStack: Plot stacked logos for single or multiple DNA, RNA and amino acid sequence. R package version 1.20.1.
Sandelin, A., Höglund, A., Lenhard, B., & Wasserman, W. W. (2003). Integrated analysis of yeast regulatory sequences for biologically linked clusters of genes. Functional & Integrative Genomics, 3(3), 125–134.
Schneider, T. D. & Stephens, R. R. (1990). A New Way to Display Consensus Sequences. Nucleic Acid Research, 18, 6097–6100.
Schneider, T. D., Stormo, G. D., Gold, L., & Ehrenfeucht, A. (1986). Information content of binding sites on nucleotide sequences. Journal of Molecular Biology, 188, 415–431.
Shannon, P. (2017). MotifDb: An Annotated Collection of Protein-DNA Binding Sequence Motifs. R package version 1.18.0.
Stojnic, R. & Diez, D. (2015). PWMEnrich: PWM enrichment analysis. R package version 4.12.0.
Tan, G. (2014). JASPAR2014: Data package for JASPAR. R package version 1.12.0.
Tan, G. (2015). JASPAR2016: Data package for JASPAR 2016. R package version 1.4.0.
Tan, G. & Lenhard, B. (2016). TFBSTools: an R/Bioconductor package for transcription factor binding site analysis. Bioinformatics, 32, 1555–1556.
Wasserman, W. W. & Sandelin, A. (2004). Applied bioinformatics for the identification of regulatory elements. Nature Reviews Genetics, 5, 276–287.
Session info¶
sessionInfo()
R version 3.4.3 (2017-11-30)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.4 LTS
Matrix products: default
BLAS: /usr/local/lib/R/lib/libRblas.so
LAPACK: /usr/local/lib/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=hu_HU.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=hu_HU.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=hu_HU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=hu_HU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 parallel stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] XtraSNPlocs.Hsapiens.dbSNP144.GRCh38_0.99.12
[2] rGADEM_2.26.0
[3] PWMEnrich_4.14.0
[4] BSgenome.Hsapiens.UCSC.hg19_1.4.0
[5] seqLogo_1.44.0
[6] JASPAR2016_1.6.0
[7] JASPAR2014_1.14.0
[8] MotifDb_1.20.0
[9] TFBSTools_1.16.0
[10] SNPlocs.Hsapiens.dbSNP150.GRCh38_0.99.20
[11] BSgenome_1.46.0
[12] rtracklayer_1.38.3
[13] Biostrings_2.46.0
[14] XVector_0.18.0
[15] GenomicRanges_1.30.3
[16] GenomeInfoDb_1.14.0
[17] IRanges_2.12.0
[18] S4Vectors_0.16.0
[19] BiocGenerics_0.24.0
loaded via a namespace (and not attached):
[1] httr_1.3.1 Biobase_2.38.0
[3] VGAM_1.0-5 bit64_0.9-7
[5] jsonlite_1.5 splines_3.4.3
[7] R.utils_2.6.0 gtools_3.5.0
[9] blob_1.1.1 GenomeInfoDbData_1.0.0
[11] Rsamtools_1.30.0 DirichletMultinomial_1.20.0
[13] pillar_1.2.1 RSQLite_2.1.0
[15] lattice_0.20-35 uuid_0.1-2
[17] digest_0.6.15 colorspace_1.3-2
[19] Matrix_1.2-13 R.oo_1.21.0
[21] plyr_1.8.4 XML_3.98-1.10
[23] pkgconfig_2.0.1 zlibbioc_1.24.0
[25] xtable_1.8-2 GO.db_3.5.0
[27] scales_0.5.0 gdata_2.18.0
[29] BiocParallel_1.12.0 tibble_1.4.2
[31] annotate_1.56.2 KEGGREST_1.18.1
[33] ggplot2_2.2.1 SummarizedExperiment_1.8.1
[35] repr_0.12.0 TFMPvalue_0.0.6
[37] lazyeval_0.2.1 splitstackshape_1.4.4
[39] magrittr_1.5 crayon_1.3.4
[41] memoise_1.1.0 poweRlaw_0.70.1
[43] evaluate_0.10.1 R.methodsS3_1.7.1
[45] CNEr_1.14.0 BiocInstaller_1.28.0
[47] data.table_1.10.4-3 tools_3.4.3
[49] hms_0.4.2 matrixStats_0.53.1
[51] stringr_1.3.0 munsell_0.4.3
[53] DelayedArray_0.4.1 AnnotationDbi_1.40.0
[55] compiler_3.4.3 evd_2.3-2
[57] caTools_1.17.1 rlang_0.2.0
[59] RCurl_1.95-4.10 pbdZMQ_0.3-2
[61] IRkernel_0.8.12.9000 bitops_1.0-6
[63] gtable_0.2.0 DBI_0.8
[65] reshape2_1.4.3 R6_2.2.2
[67] GenomicAlignments_1.14.2 bit_1.1-12
[69] readr_1.1.1 stringi_1.1.7
[71] IRdisplay_0.4.4 Rcpp_0.12.16
[73] png_0.1-7