NGS¶
https://www.youtube.com/watch?v=9vNBYyHedLg
https://www.youtube.com/watch?v=ToKUGz_YhC4
https://www.youtube.com/watch?v=9YxExTSwgPM
Short Reads¶
SRA¶
https://www.ncbi.nlm.nih.gov/sra/docs/
példa: SRR1660257: https://www.ncbi.nlm.nih.gov/sra/?term=SRR1660257
https://www.ncbi.nlm.nih.gov/geo/
%%bash
export PATH=$PATH:/usr/local/ncbi/sra-tools/bin
mkdir gyak06
cd gyak06
fastq-dump SRR1660257
Read 1015290 spots for SRR1660257
Written 1015290 spots for SRR1660257
%%bash
cd gyak06
ls -lh
total 267M
-rw-rw-r-- 1 sn sn 267M ápr 3 11:18 SRR1660257.fastq
%%bash
cd gyak06
head SRR1660257.fastq
@SRR1660257.1 1 length=100
GTAATCTTAGATTCAAGTAATCCATATAATTAAATCCTGGTTTCATTGATTTGAGGCTGAGCTTTTCCATGAAGCAATCTGAAGAAAAACTAAAAGACAA
+SRR1660257.1 1 length=100
CBCFFFFFHHHHHJJIJHIJJJJJJJJJJJJJJJJJJJJJGIJJJJJJJJJJJIJJJJJIJJJJJJJJJJJJJJJIJJJJJJHHGHHHFFFFCEECDDDD
@SRR1660257.2 2 length=100
CAAGAGCACTGACTTCCTGGACCCCGCCACCACAACAAGTCCCCAAAACCACAGCGAGACCGCTGGCAACAACAACACTCATCACCAAGATACCGGAGAA
+SRR1660257.2 2 length=100
CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJJJIJJJJJIJJJJJJJIJJJJJJHHFFDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDD
@SRR1660257.3 3 length=100
GCCCAAGATTGATCGAGGTTGGGTATGTGTTTTTCAGCTTCAAGATGGTAAAACACTTGGACTCAAAATTTGAGCCAATCTCCCTTCCCTCCGAAAGAGG
FASTQ¶
https://en.wikipedia.org/wiki/FASTQ_format
Minden readhez négy sor tartozik: - az első sorban van a read azonosítója, illetve lehet még hozzá tartozó leírás - a második sorban van a read nyers nukleotidsora - a harmadik + jellel kezdődő sor a jel után lehet üres vagy a read azonosítóját is tartalmazhatja - a negyedik sorban az egyes nukleotidok leolvasási minőségére vonatkozó kódolás (részletek 74. lapon: Csaba Ortutay, Zsuzsanna Ortutay: Molecular Data Analysis Using R. 2017, Wiley-Blackwell, ISBN: 978-1-119-16502-6. https://www.wiley.com/en-hu/Molecular+Data+Analysis+Using+R-p-9781119165026)
BAM-ban tárolva: https://gatkforums.broadinstitute.org/gatk/discussion/5990/what-is-ubam-and-why-is-it-better-than-fastq-for-storing-unmapped-sequence-data
CSFASTA¶
ABi SOLiD sequencer: http://cutadapt.readthedocs.io/en/stable/colorspace.html
CSFASTA-file
# Title: s0205_20110422_FRAG_BC_miRNA_MeDIP
>1_5_224_F3
T.222200232103..132..030.020..000.00
>1_5_656_F3
T.31311231.331..233..122.122..122.10
>1_5_1005_F3"
T.122221311011..212..312.033..303.32
| A | C | G | T | |
|---|---|---|---|---|
| A | 0 | 1 | 2 | 3 |
| C | 1 | 0 | 3 | 2 |
| G | 2 | 3 | 0 | 1 |
| T | 3 | 2 | 1 | 0 |
QUAL-file
# Title: s0205_20110422_FRAG_BC_miRNA_MeDIP
>1_5_224_F3"
-1 27 28 30 32 21 31 30 31 27 4 28 31 -1 -1 17 28 31 -1 -1 24 30 25 -1 22 25 21 -1 -1 30 28 21 -1 27 31
>1_5_656_F3"
-1 33 4 31 29 16 33 4 32 -1 20 33 4 -1 -1 25 31 10 -1 -1 12 25 5 -1 22 4 7 -1 -1 5 4 7 -1 4 4
>1_5_1005_F3"
-1 31 20 33 32 32 30 32 31 26 26 25 17 -1 -1 30 26 6 -1 -1 27 23 28 -1 25 31 10 -1 -1 29 14 19 -1 33 33
%%bash
cd gyak06
fastq-dump -X 10000 --split-files SRR1972739
Read 10000 spots for SRR1972739
Written 10000 spots for SRR1972739
library(ShortRead)
library(seqTools)
library(qrqc)
library(BiocParallel)
setwd('gyak06')
fajlom = 'SRR1660257.fastq'
readek = readFastq(fajlom)
slotNames(readek)
- 'quality'
- 'sread'
- 'id'
id(readek)
A BStringSet instance of length 1015290
width seq
[1] 25 SRR1660257.1 1 length=100
[2] 25 SRR1660257.2 2 length=100
[3] 25 SRR1660257.3 3 length=100
[4] 25 SRR1660257.4 4 length=100
[5] 25 SRR1660257.5 5 length=100
... ... ...
[1015286] 37 SRR1660257.1015286 1015286 length=100
[1015287] 37 SRR1660257.1015287 1015287 length=100
[1015288] 37 SRR1660257.1015288 1015288 length=100
[1015289] 37 SRR1660257.1015289 1015289 length=100
[1015290] 37 SRR1660257.1015290 1015290 length=100
sread(readek)
A DNAStringSet instance of length 1015290
width seq
[1] 100 GTAATCTTAGATTCAAGTAATCCATATAATT...GAAGCAATCTGAAGAAAAACTAAAAGACAA
[2] 100 CAAGAGCACTGACTTCCTGGACCCCGCCACC...AACAACACTCATCACCAAGATACCGGAGAA
[3] 100 GCCCAAGATTGATCGAGGTTGGGTATGTGTT...TGAGCCAATCTCCCTTCCCTCCGAAAGAGG
[4] 100 TAATAATCAGATCTGCGAACCGGTAGAGTTT...CAATAGAAATTTAAACAGTGAGTGGAGACA
[5] 100 CTTAGACATCAAAAATTCTTCCTGTTTTCGT...ATCCCATTGTTCCATGCTCATTCACTGATG
... ... ...
[1015286] 100 CAGATGATGAAGAGCAGGACAGGGACGGAAC...ATACAGAGATCACTCTGAAAAGAAAGAACT
[1015287] 100 CTGGAAGTTCATAAGAATTTTCTTTTCCTGA...TATTGTTGGAGTTGCTTCTCAGCCTCAGTG
[1015288] 100 ATCTTCCAAGATGCTGCTCCACCTGTCATCC...AAAGCTTGCGTCCAGTCCCACCATCGCCCA
[1015289] 100 AGAGCCACAACTGAGCTACGCACCTTTTCAA...GGGGCGGCACATGCCACATTCTGGGACCGG
[1015290] 100 GACGAGGACACTAAGCCGGTGCCTAATAGAT...GGCATATAGAGGGCAGACAGACACAATCCG
kval = quality(readek)
kval
class: FastqQuality
quality:
A BStringSet instance of length 1015290
width seq
[1] 100 CBCFFFFFHHHHHJJIJHIJJJJJJJJJJJJ...JJJJJIJJJJJJHHGHHHFFFFCEECDDDD
[2] 100 CCCFFFFFHHHHHJJJJJJJJJJJJJJJJJJ...DDDDDDDDDDDDDDDDDDDDDDDDDDDDDD
[3] 100 CCCFFFFFHHHHHJJIJJEGIJJCFGICFFG...HHFFFFECEEEEEDDDDDDDDDDDDDDDDB
[4] 100 CCCFFFFFHHHHHJJJJGJJJJJGHIJJHIJ...EEEEEEDCCDDDDEDDDDCCDDACDDDDDD
[5] 100 CCCFFFFFHHHHHJJJJJJJJJJJJJJJJIJ...JJGHHHHFFFFFFFECCCEEEEECCDDDDD
... ... ...
[1015286] 100 C@CFFFFFHHHHHJBHIIIJJIIIGIGGIIJ...DDDEEDDDCCDDDDDDDDDDDDDDDDDDDC
[1015287] 100 ?@@FBDBBFHHBBGBECFFHIBEHEHDHHEG...CH>DF@@;;(..;AC@C>A@;C>9<ACC@:
[1015288] 100 CCCFFFFFHHHHHJJJJJJJJIJJJHIIJJI...DEEDBCDDDDDDBDCCDDDDDDDCDDDDDD
[1015289] 100 ;@@DDDDDHHFDCCGBHGI@GHFFCHHGIEC...A92=;59<@><@AACCCBAACDACCBBB<5
[1015290] 100 1=@D4=A########################...##############################
PHRED minőségi pontszám:
\(Q_{PHRED} = -10 \times log_{10} P\)
Ennek valószínűséggé való átalakítása:
\(P = 10^{-Q_{PHRED}/10}\)
Néhány minőségi érték a döntésekhez:
| \(Q_{PHRED}\) | hiba | megbízhatóság | |
|---|---|---|---|
| 10 | 10% | (1/10) | 90% |
| 20 | 1% | (1/100) | 99% |
| 30 | 0.1% | (1/1000) | 99.9% |
| 40 | 0.01% | (1/10000) | 99.99% |
Solexa:
\(Q_{Solexa}=-10\times log_{10}\left(\frac{P}{1-P}\right)\)
Átváltások:
\(Q_{PHRED}=-10\times log_{10}\left(10^{Q_{Solexa}/10}+1\right)\)
\(Q_{Solexa}=-10\times log_{10}\left(10^{Q_{PHRED}/10}-1\right)\)
Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM The sanger FASTQ file format for sequences with quality scores and the Solexa/Illumina FASTQ variants. Nucleic Acids Research 2010 Vol.38 No.6 1767-1771
phredTable()
| ascii | phred | char |
|---|---|---|
| 33 | 0 | ! |
| 34 | 1 | " |
| 35 | 2 | # |
| 36 | 3 | $ |
| 37 | 4 | % |
| 38 | 5 | & |
| 39 | 6 | ' |
| 40 | 7 | ( |
| 41 | 8 | ) |
| 42 | 9 | * |
| 43 | 10 | + |
| 44 | 11 | , |
| 45 | 12 | - |
| 46 | 13 | . |
| 47 | 14 | / |
| 48 | 15 | 0 |
| 49 | 16 | 1 |
| 50 | 17 | 2 |
| 51 | 18 | 3 |
| 52 | 19 | 4 |
| 53 | 20 | 5 |
| 54 | 21 | 6 |
| 55 | 22 | 7 |
| 56 | 23 | 8 |
| 57 | 24 | 9 |
| 58 | 25 | : |
| 59 | 26 | ; |
| 60 | 27 | < |
| 61 | 28 | = |
| 62 | 29 | > |
| ⋮ | ⋮ | ⋮ |
| 97 | 64 | a |
| 98 | 65 | b |
| 99 | 66 | c |
| 100 | 67 | d |
| 101 | 68 | e |
| 102 | 69 | f |
| 103 | 70 | g |
| 104 | 71 | h |
| 105 | 72 | i |
| 106 | 73 | j |
| 107 | 74 | k |
| 108 | 75 | l |
| 109 | 76 | m |
| 110 | 77 | n |
| 111 | 78 | o |
| 112 | 79 | p |
| 113 | 80 | q |
| 114 | 81 | r |
| 115 | 82 | s |
| 116 | 83 | t |
| 117 | 84 | u |
| 118 | 85 | v |
| 119 | 86 | w |
| 120 | 87 | x |
| 121 | 88 | y |
| 122 | 89 | z |
| 123 | 90 | { |
| 124 | 91 | | |
| 125 | 92 | } |
| 126 | 93 | ~ |
m = as(kval, 'matrix')
m[1,]
- 34
- 33
- 34
- 37
- 37
- 37
- 37
- 37
- 39
- 39
- 39
- 39
- 39
- 41
- 41
- 40
- 41
- 39
- 40
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 38
- 40
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 40
- 41
- 41
- 41
- 41
- 41
- 40
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 41
- 40
- 41
- 41
- 41
- 41
- 41
- 41
- 39
- 39
- 38
- 39
- 39
- 39
- 37
- 37
- 37
- 37
- 34
- 36
- 36
- 34
- 35
- 35
- 35
- 35
Readek minőségellenőrzése¶
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2752612/
FastQC: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
https://www.youtube.com/watch?v=bz93ReOv87Y
%%bash
# export PATH=$PATH:/usr/local/ncbi/sra-tools/bin
mkdir gyak07
cd gyak07
fastq-dump SRR1660259
ls -lh
Read 9130 spots for SRR1660259
Written 9130 spots for SRR1660259
total 2,4M
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
library(fastqcr)
setwd('gyak07')
fastqc()
minosegi.fajl = 'FASTQC/SRR1660259_fastqc.zip'
qc_report(
qc.path = minosegi.fajl,
template = 'minta_jelentes.Rmd',
result.file ='osszefoglalo',
interpret = TRUE,
experiment = 'minőségellenőrzési példa',
preview = FALSE
)
processing file: minta_jelentes.Rmd
|. | 2%
inline R code fragments
|.. | 3%
label: unnamed-chunk-1 (with options)
List of 1
$ echo: logi FALSE
|... | 5%
ordinary text without R code
|.... | 6%
label: unnamed-chunk-2 (with options)
List of 1
$ echo: logi FALSE
|..... | 8%
inline R code fragments
|...... | 10%
label: unnamed-chunk-3 (with options)
List of 1
$ echo: logi TRUE
Attaching package: 'dplyr'
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
|....... | 11%
ordinary text without R code
|........ | 13%
label: unnamed-chunk-4
Reading: FASTQC/SRR1660259_fastqc.zip
|......... | 14%
ordinary text without R code
|.......... | 16%
label: summary (with options)
List of 3
$ fig.width : num 5
$ fig.height: num 4.5
$ fig.align : chr "center"
|........... | 17%
ordinary text without R code
|............ | 19%
label: basic-statistics (with options)
List of 3
$ fig.width : num 5
$ fig.height: num 3
$ fig.align : chr "center"
|............. | 21%
ordinary text without R code
|.............. | 22%
label: per-base-sequence-quality (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|............... | 24%
ordinary text without R code
|................. | 25%
label: unnamed-chunk-5 (with options)
List of 2
$ type : chr "warning"
$ engine: chr "block"
|.................. | 27%
ordinary text without R code
|................... | 29%
label: unnamed-chunk-6 (with options)
List of 2
$ type : chr "block"
$ engine: chr "block"
|.................... | 30%
ordinary text without R code
|..................... | 32%
label: per-sequence-quality-scores (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|...................... | 33%
ordinary text without R code
|....................... | 35%
label: unnamed-chunk-7 (with options)
List of 2
$ type : chr "warning"
$ engine: chr "block"
|........................ | 37%
ordinary text without R code
|......................... | 38%
label: unnamed-chunk-8 (with options)
List of 2
$ type : chr "block"
$ engine: chr "block"
|.......................... | 40%
ordinary text without R code
|........................... | 41%
label: per-base-sequence-content (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|............................ | 43%
ordinary text without R code
|............................. | 44%
label: unnamed-chunk-9 (with options)
List of 2
$ type : chr "notice"
$ engine: chr "block"
|.............................. | 46%
ordinary text without R code
|............................... | 48%
label: unnamed-chunk-10 (with options)
List of 2
$ type : chr "warning"
$ engine: chr "block"
|................................ | 49%
ordinary text without R code
|................................. | 51%
label: unnamed-chunk-11 (with options)
List of 2
$ type : chr "block"
$ engine: chr "block"
|.................................. | 52%
ordinary text without R code
|................................... | 54%
label: per-sequence-GC-content (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|.................................... | 56%
ordinary text without R code
|..................................... | 57%
label: unnamed-chunk-12 (with options)
List of 2
$ type : chr "success"
$ engine: chr "block"
|...................................... | 59%
ordinary text without R code
|....................................... | 60%
label: per-base-N-content (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|........................................ | 62%
ordinary text without R code
|......................................... | 63%
label: unnamed-chunk-13 (with options)
List of 2
$ type : chr "warning"
$ engine: chr "block"
|.......................................... | 65%
ordinary text without R code
|........................................... | 67%
label: unnamed-chunk-14 (with options)
List of 2
$ type : chr "block"
$ engine: chr "block"
|............................................ | 68%
ordinary text without R code
|............................................. | 70%
label: sequence-length-distribution (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
geom_path: Each group consists of only one observation. Do you need to adjust the group aesthetic?
|.............................................. | 71%
ordinary text without R code
|............................................... | 73%
label: sequence-duplication-levels (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|................................................ | 75%
ordinary text without R code
|.................................................. | 76%
label: unnamed-chunk-15 (with options)
List of 2
$ type : chr "warning"
$ engine: chr "block"
|................................................... | 78%
ordinary text without R code
|.................................................... | 79%
label: unnamed-chunk-16 (with options)
List of 2
$ type : chr "block"
$ engine: chr "block"
|..................................................... | 81%
ordinary text without R code
|...................................................... | 83%
label: Overrepresented-sequences (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|....................................................... | 84%
ordinary text without R code
|........................................................ | 86%
label: unnamed-chunk-17 (with options)
List of 2
$ type : chr "warning"
$ engine: chr "block"
|......................................................... | 87%
ordinary text without R code
|.......................................................... | 89%
label: unnamed-chunk-18 (with options)
List of 2
$ type : chr "block"
$ engine: chr "block"
|........................................................... | 90%
ordinary text without R code
|............................................................ | 92%
label: adapter-content (with options)
List of 3
$ fig.width : num 4
$ fig.height: num 3.5
$ fig.align : chr "center"
|............................................................. | 94%
ordinary text without R code
|.............................................................. | 95%
label: unnamed-chunk-19 (with options)
List of 2
$ type : chr "warning"
$ engine: chr "block"
|............................................................... | 97%
ordinary text without R code
|................................................................ | 98%
label: unnamed-chunk-20 (with options)
List of 2
$ type : chr "block"
$ engine: chr "block"
|.................................................................| 100%
ordinary text without R code
output file: minta_jelentes.knit.md
/usr/bin/pandoc +RTS -K512m -RTS minta_jelentes.utf8.md --to html4 --from markdown+autolink_bare_uris+ascii_identifiers+tex_math_single_backslash --output /home/sn/gyak07/osszefoglalo.html --smart --email-obfuscation none --self-contained --standalone --section-divs --table-of-contents --toc-depth 3 --variable toc_float=1 --variable toc_selectors=h1,h2,h3 --variable toc_smooth_scroll=1 --variable toc_print=1 --template /usr/local/lib/R/library/rmarkdown/rmd/h/default.html --no-highlight --variable highlightjs=1 --variable 'theme:bootstrap' --include-in-header /tmp/Rtmp25z3BX/rmarkdown-str13c555fdd057.html --mathjax --variable 'mathjax-url:https://mathjax.rstudio.com/latest/MathJax.js?config=TeX-AMS-MML_HTMLorMML'
Output created: osszefoglalo.html
--------------------------
Output file: /home/sn/gyak07/osszefoglalo.html
--------------------------
Gyenge minőségű nukleotidok, readek kiszűrése¶
trimming, filtering
library(seqTools)
fajlom = 'SRR1660259.fastq'
trimFastq(fajlom,
outfile = 'marad.fq.gz',
discard = 'kimarad.fq.gz',
qualDiscard = 10, # All reads which contain one or more phred scores < qualDiscard will be discarded.
fixTrimLeft = 0, # Prefix of this size will be trimmed.
fixTrimRight = 0, # Suffix of this size will be trimmed.
qualTrimLeft = 30, # Prefix where all phred scores are < qualTrimLeft will be trimmed.
qualTrimRight = 30, # Suffix where all phred scores are < qualTrimRight will be trimmed.
minSeqLen = 50 # All reads where sequence length after (fixed and quality based) trimming is < minSeqLen will be discarded.
)
Loading required package: zlibbioc
[trimFastq] 7.843 records written to outfile.
[trimFastq] 1.287 records written to discard.
Readek illesztése referencia-genomra¶
%%bash
cd gyak07
export PATH=$PATH:/home/bioinfo/edirect
efetch -db=nuccore -format=fasta -id=AF086833 > ebola1976.fa
ls -lh
total 4,6M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
%%bash
cd gyak07
head ebola1976.fa
>AF086833.2 Ebola virus - Mayinga, Zaire, 1976, complete genome
CGGACACACAAAAAGAAAGAAGAATTTTTAGGATCTTTTGTGTGCGAATAACTATGAGGAAGATTAATAA
TTTTCCTCTCATTGAAATTTATATCGGAATTTAAATTGAAATTGTTACTGTAATCACACCTGGTTTGTTT
CAGAGCCACATCACAAAGATAGAGAACAACCTAGGTCTCCGAAGGGAGCAAGGGCATCAGTGTGCTCAGT
TGAAAATCCCTTGTCAACACCTAGGTCTTATCACATCACAAGTTCCACCTCAGACTCTGCAGGGTGATCC
AACAACCTTAATAGAAACATTATTGTTAAAGGACAGCATTAGTTCACAGTCAAACAAGCAAGATTGAGAA
TTAACCTTGGTTTTGAACTTGAACACTTAGGGGATTGAAGATTCAACAACCCTAAAGCTTGGGGTAAAAC
ATTGGAAATAGTTAAAAGACAAATTGCTCGGAATCACAAAATTCCGAGTATGGATTCTCGTCCTCAGAAA
ATCTGGATGGCGCCGAGTCTCACTGAATCTGACATGGATTACCACAAGATCTTGACAGCAGGTCTGTCCG
TTCAACAGGGGATTGTTCGGCAAAGAGTCATCCCAGTGTATCAAGTAAACAATCTTGAAGAAATTTGCCA
%%bash
cd gyak07
export PATH=$PATH:/home/bioinfo/bwa
bwa index -p Ebola ebola1976.fa
ls -lh
total 4,6M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
-rw-rw-r-- 1 sn sn 10 ápr 3 11:34 Ebola.amb
-rw-rw-r-- 1 sn sn 86 ápr 3 11:34 Ebola.ann
-rw-rw-r-- 1 sn sn 19K ápr 3 11:34 Ebola.bwt
-rw-rw-r-- 1 sn sn 4,7K ápr 3 11:34 Ebola.pac
-rw-rw-r-- 1 sn sn 9,4K ápr 3 11:34 Ebola.sa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
[bwa_index] Pack FASTA... 0.00 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 0.00 seconds elapse.
[bwa_index] Update BWT... 0.00 sec
[bwa_index] Pack forward-only FASTA... 0.00 sec
[bwa_index] Construct SA from BWT and Occ... 0.00 sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa index -p Ebola ebola1976.fa
[main] Real time: 0.427 sec; CPU: 0.009 sec
%%bash
cd gyak07
export PATH=$PATH:/home/bioinfo/bwa
bwa mem Ebola SRR1660259.fastq > illesztes01.sam
ls -lh
total 7,1M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
-rw-rw-r-- 1 sn sn 10 ápr 3 11:34 Ebola.amb
-rw-rw-r-- 1 sn sn 86 ápr 3 11:34 Ebola.ann
-rw-rw-r-- 1 sn sn 19K ápr 3 11:34 Ebola.bwt
-rw-rw-r-- 1 sn sn 4,7K ápr 3 11:34 Ebola.pac
-rw-rw-r-- 1 sn sn 9,4K ápr 3 11:34 Ebola.sa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 2,5M ápr 3 11:34 illesztes01.sam
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 9130 sequences (911661 bp)...
[M::mem_process_seqs] Processed 9130 reads in 0.323 CPU sec, 0.332 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem Ebola SRR1660259.fastq
[main] Real time: 0.418 sec; CPU: 0.347 sec
Sequence Alignment Map (SAM) fájl¶
Az illesztés eredményét tartalmazó tabulátorral osztott fájl (CSV), az egyes részek nem azonos oszlopszámúak (https://samtools.github.io/hts-specs/SAMv1.pdf).
%%bash
cd gyak07
head illesztes01.sam
@SQ SN:AF086833.2 LN:18959
@PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem Ebola SRR1660259.fastq
SRR1660259.1 16 AF086833.2 7706 60 100M * 0 0 GGGTTGAGACAGCTGGCCAACGAGACGACTCAAGCTCTTCAACTGTTCCTGAGAGCCACAACTGAGCTACGCACCTTTTCAATCCTCAACCGTAAGGCAA >@CCCA>@5C>??3CA?9==CAAACA@@FEDCEHEHHGEHGEGFEGBIIHEFEIGHF?BFDGHHCIGGHBGCHIGIIIGGHFC+GBAFFHDHFFFFF@@@ NM:i:0 MD:Z:100 AS:i:100 XS:i:0
SRR1660259.2 0 AF086833.2 814 60 100M * 0 0 GCGCCTTGAGGAATTGCTGCCAGCAGTATCTAGTGGAAAAAACATTAAGAGAACACTTGCTGCCATGCCGGAAGAGGAGACAACTGAAGCTAATGCCGGT CCCFFFFFHHHHHJJJJJJJJJJJJJHIJJJJJHHIJJJJIJJJJJIJJJJGIIJJJJJJJJJJJJHHHHFDDDDDDDDDDDDDDDDDDDDDDDDDDDDB NM:i:0 MD:Z:100 AS:i:100 XS:i:0
SRR1660259.3 0 AF086833.2 11530 60 83M17S * 0 0 GTCTTTCCGTGTTTAAGATGGAGCAGTTGAAATTCTTCCTCTTGATATTAAATGGCTACACAACATACCCAATACCCAGACGCGTGAGCAAGGGCGAGGA @CCDF?EFHHHDHJJJJEIJJIGGIJHIIJIIJJJGGHJJFGIIDGIJIIIHIIGEGIGGHJIJIIGHHHJGIG:CAEEDFF@DDDDDDDDDDDDDDD@9 NM:i:2 MD:Z:14T5A62 AS:i:73 XS:i:0
SRR1660259.4 16 AF086833.2 1065 60 100M * 0 0 TGATTTTCCGTTTGATGCGAACAAATTTTCTGATCAAATTTCTCCTAATACACCAAGGGATGCACATGGTTGCCGGGCATGATGCCAACGATGCTGTGAT DDDDDDDDDDDCDDDDDDDDCEEEEEFFFFFHHHHHHHGIHJJJIIJIHCGCJJIIJJJJJJIGJJJJIJJJJJIJJJJJJJJJJJJHHHHHFFFFFCCC NM:i:0 MD:Z:100 AS:i:100 XS:i:0
SRR1660259.5 0 AF086833.2 11510 60 100M * 0 0 TAAGAAAAACTGCTTATTGGGTCTTTCCGTGTTTTAGATGAAGCAGTTGAAATTCTTCCTCTTGATATTAAATGGCTACACAACATACCCAATACCCAGA CCCFFFFFHHHHHJJJJJJJJFHJJJJJJGHHJJJJJIJJIJJJJJIIJJJJJIJIIJJJJJJJJJJJJJIJJIJIHHGHHFFFFEEEEEDDDDDDDDDD NM:i:0 MD:Z:100 AS:i:100 XS:i:0
SRR1660259.6 0 AF086833.2 2660 60 100M * 0 0 TTCATGGCAATCCTGCAACATCATCAGTGAATGAGCATGGAACAATGGGATGATTCAACCGACAAATAGCTAACATTAAGTAGTCAAGGAACGAAAACAG CCCFFFFFHHHHHJJJJJJJJJJJJJJHIJJJJJJJJJJJIJJJJJJJJJJJIJJJJJJJJJJJHHHHHFFFFFFEEEEEDEEFEEDDDDDDDDDDDDDD NM:i:0 MD:Z:100 AS:i:100 XS:i:0
SRR1660259.7 16 AF086833.2 7235 60 100M * 0 0 GAGGCAACTCAAGTTGAACAACATCCCCGCAGAACAGACAACGACAGCACAGCCTCCGACACTCCCTCTGCCACGACCGCAGCCGGACCCCCAAAAGCAG ACDDDDCC@CCDDEDCDCCCC<<<5&B@ACCAC@:C>?<BDDC@CCA>8?DDDDDDDEA>5<<FHC;HGJIIIIGFJJJJJJJJJJJHHHFHFFFFFCCC NM:i:1 MD:Z:25A74 AS:i:95 XS:i:0
SRR1660259.8 16 AF086833.2 3969 60 100M * 0 0 ACAAAAAGAGTTCCAATCTTCCAAGATGCTGCTCCACCTGTCATCCACATCCGCTCTCGAGGTGACATTCCCCGAGCTTGCCAGAAAAGCTTGCGTCCAG <BDDDDDDDDDDDDDDBCDDDDDDCDDBDBDDBDCCDDDDDDDDDDCDDDFHHJJJIJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJHHHHHFFFFFCCC NM:i:0 MD:Z:100 AS:i:100 XS:i:0
Fejléc:
@SQ SN:AF086833.2 LN:18959
@PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem ../genomes/Ebola SRR1660257.fastq
Illesztési eredmény 1. sora:
SRR1660257.1 16 AF086833.2 8058 60 100M * 0 0TTGTCTTTTAGTTTTTCTTCAGATTGCTTCATGGAAAAGCTCAGCCTCAAATCAATGAAACCAGGATTTAATTATATGGATTACTTGAATCTAAGATTAC DDDDCEECFFFFHHHGHHJJJJJJIJJJJJJJJJJJJJJJIJJJJJIJJJJJJJJJJJIGJJJJJJJJJJJJJJJJJJJJJIHJIJJHHHHHFFFFFCBC NM:i:0 MD:Z:100 AS:i:100XS:i:0
Kötelező elemei:
QNAME = SRR1660257.1 # Query template NAME
FLAG = 16 # bitwise FLAG; 16=SEQ being reverse complemented
RNAME = AF086833.2 # Reference sequence NAME
POS = 8058 # 1-based leftmost mapping POSition
MAPQ = 60 # MAPping Quality (https://genome.sph.umich.edu/wiki/Mapping_Quality_Scores)
CIGAR = 100M # CIGAR string
RNEXT = * # Ref. name of the mate/next read
PNEXT = 0 # Position of the mate/next read
TLEN = 0 # observed Template LENgth
SEQ = TTGTCTTTTAGTTTTTCTTCAGATTGCTTCATGGAAAAGCTCAGCCTCAAATCAATGAAACCAGGATTTAATTATATGGATTACTTGAATCTAAGATTAC
QUAL = DDDDCEECFFFFHHHGHHJJJJJJIJJJJJJJJJJJJJJJIJJJJJIJJJJJJJJJJJIGJJJJJJJJJJJJJJJJJJJJJIHJIJJHHHHHFFFFFCBC
Opcionális elemei (http://samtools.github.io/hts-specs/SAMtags.pdf):
NM:i:0 # Edit distance to the reference, including ambiguous bases but excluding clipping
MD:Z:100 # String for mismatching positions, the field ought to match the CIGAR string.
AS:i:100 # Alignment score generated by aligner
XS:i:0 # Reserved for end users
A MAPQ azt fejezi ki PHRED-pontszámmal, hogy a read milyen valószínűséggel lett hibásan illesztve.
CIGAR string értelmezéséhez:
M alignment match (can be a sequence match or mismatch)
I insertion to the reference
D deletion from the reference
N skipped region from the reference
S soft clipping (clipped sequences present in SEQ)
H hard clipping (clipped sequences NOT present in SEQ)
P padding (silent deletion from padded reference)
= sequence match
X sequence mismatch
Binary Alignment Map (BAM) fájl¶
Bináris, tömörített SAM-fájl. A kisebb mérete mellett nagy előnye, hogy lekérdezhető, kisebb részletek kiolvashatók belőle anélkül, hogy a teljes állományt be kellene tölteni a memóriába.
%%bash
cd gyak07
samtools view -Sb illesztes01.sam > illesztes01.bam
ls -lh
total 7,7M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
-rw-rw-r-- 1 sn sn 10 ápr 3 11:34 Ebola.amb
-rw-rw-r-- 1 sn sn 86 ápr 3 11:34 Ebola.ann
-rw-rw-r-- 1 sn sn 19K ápr 3 11:34 Ebola.bwt
-rw-rw-r-- 1 sn sn 4,7K ápr 3 11:34 Ebola.pac
-rw-rw-r-- 1 sn sn 9,4K ápr 3 11:34 Ebola.sa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 639K ápr 3 11:39 illesztes01.bam
-rw-rw-r-- 1 sn sn 2,5M ápr 3 11:34 illesztes01.sam
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
%%bash
cd gyak07
samtools sort illesztes01.bam > illesztes01_sorted.bam
samtools index illesztes01_sorted.bam
ls -lh
total 8,1M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
-rw-rw-r-- 1 sn sn 10 ápr 3 11:34 Ebola.amb
-rw-rw-r-- 1 sn sn 86 ápr 3 11:34 Ebola.ann
-rw-rw-r-- 1 sn sn 19K ápr 3 11:34 Ebola.bwt
-rw-rw-r-- 1 sn sn 4,7K ápr 3 11:34 Ebola.pac
-rw-rw-r-- 1 sn sn 9,4K ápr 3 11:34 Ebola.sa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 639K ápr 3 11:39 illesztes01.bam
-rw-rw-r-- 1 sn sn 2,5M ápr 3 11:34 illesztes01.sam
-rw-rw-r-- 1 sn sn 449K ápr 3 11:39 illesztes01_sorted.bam
-rw-rw-r-- 1 sn sn 128 ápr 3 11:39 illesztes01_sorted.bam.bai
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
# TERMINÁLBAN !!!
cd gyak07
samtools tview illesztes01_sorted.bam
samtools tview illesztes01_sorted.bam ebola1976.fa
- ?: súgó ablakot nyit
- .: forward illesztésű, a referenciával megegyező nukleotid
- ,: reverse illesztésű, a referenciával megegyező nukleotid
- ACGT: forward illesztésű, referenciától eltérő nukleotid
- acgt: reverse illesztésű, referenciától eltérő nukleotid
- *: törölt bázis
%%bash
cd gyak07
samtools view -Sb -F 4 illesztes01.sam > illesztes01_van.bam
samtools view -Sb -f 4 illesztes01.sam > illesztes01_nincs.bam
ls -lh
total 8,8M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
-rw-rw-r-- 1 sn sn 10 ápr 3 11:34 Ebola.amb
-rw-rw-r-- 1 sn sn 86 ápr 3 11:34 Ebola.ann
-rw-rw-r-- 1 sn sn 19K ápr 3 11:34 Ebola.bwt
-rw-rw-r-- 1 sn sn 4,7K ápr 3 11:34 Ebola.pac
-rw-rw-r-- 1 sn sn 9,4K ápr 3 11:34 Ebola.sa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 639K ápr 3 11:39 illesztes01.bam
-rw-rw-r-- 1 sn sn 354 ápr 3 11:41 illesztes01_nincs.bam
-rw-rw-r-- 1 sn sn 2,5M ápr 3 11:34 illesztes01.sam
-rw-rw-r-- 1 sn sn 449K ápr 3 11:39 illesztes01_sorted.bam
-rw-rw-r-- 1 sn sn 128 ápr 3 11:39 illesztes01_sorted.bam.bai
-rw-rw-r-- 1 sn sn 638K ápr 3 11:41 illesztes01_van.bam
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
%%bash
cd gyak07
export PATH=$PATH:/home/bioinfo/bwa
bwa mem Ebola SRR1660259.fastq | samtools view -Sb -F 4 > illesztes02_van.bam
ls -lh
total 9,4M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
-rw-rw-r-- 1 sn sn 10 ápr 3 11:34 Ebola.amb
-rw-rw-r-- 1 sn sn 86 ápr 3 11:34 Ebola.ann
-rw-rw-r-- 1 sn sn 19K ápr 3 11:34 Ebola.bwt
-rw-rw-r-- 1 sn sn 4,7K ápr 3 11:34 Ebola.pac
-rw-rw-r-- 1 sn sn 9,4K ápr 3 11:34 Ebola.sa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 639K ápr 3 11:39 illesztes01.bam
-rw-rw-r-- 1 sn sn 354 ápr 3 11:41 illesztes01_nincs.bam
-rw-rw-r-- 1 sn sn 2,5M ápr 3 11:34 illesztes01.sam
-rw-rw-r-- 1 sn sn 449K ápr 3 11:39 illesztes01_sorted.bam
-rw-rw-r-- 1 sn sn 128 ápr 3 11:39 illesztes01_sorted.bam.bai
-rw-rw-r-- 1 sn sn 638K ápr 3 11:41 illesztes01_van.bam
-rw-rw-r-- 1 sn sn 638K ápr 3 11:41 illesztes02_van.bam
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 9130 sequences (911661 bp)...
[M::mem_process_seqs] Processed 9130 reads in 0.325 CPU sec, 0.344 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem Ebola SRR1660259.fastq
[main] Real time: 0.484 sec; CPU: 0.345 sec
%%bash
cd gyak07
export PATH=$PATH:/home/bioinfo/bwa
bwa mem Ebola SRR1660259.fastq | samtools view -Sb -F 4 | samtools sort > illesztes03_van_sorted.bam
samtools index illesztes03_van_sorted.bam
ls -lh
total 9,8M
-rw-rw-r-- 1 sn sn 19K ápr 3 11:33 ebola1976.fa
-rw-rw-r-- 1 sn sn 10 ápr 3 11:34 Ebola.amb
-rw-rw-r-- 1 sn sn 86 ápr 3 11:34 Ebola.ann
-rw-rw-r-- 1 sn sn 19K ápr 3 11:34 Ebola.bwt
-rw-rw-r-- 1 sn sn 4,7K ápr 3 11:34 Ebola.pac
-rw-rw-r-- 1 sn sn 9,4K ápr 3 11:34 Ebola.sa
drwxrwxr-x 2 sn sn 4,0K ápr 3 11:29 FASTQC
-rw-rw-r-- 1 sn sn 639K ápr 3 11:39 illesztes01.bam
-rw-rw-r-- 1 sn sn 354 ápr 3 11:41 illesztes01_nincs.bam
-rw-rw-r-- 1 sn sn 2,5M ápr 3 11:34 illesztes01.sam
-rw-rw-r-- 1 sn sn 449K ápr 3 11:39 illesztes01_sorted.bam
-rw-rw-r-- 1 sn sn 128 ápr 3 11:39 illesztes01_sorted.bam.bai
-rw-rw-r-- 1 sn sn 638K ápr 3 11:41 illesztes01_van.bam
-rw-rw-r-- 1 sn sn 638K ápr 3 11:41 illesztes02_van.bam
-rw-rw-r-- 1 sn sn 448K ápr 3 11:42 illesztes03_van_sorted.bam
-rw-rw-r-- 1 sn sn 128 ápr 3 11:42 illesztes03_van_sorted.bam.bai
-rw-rw-r-- 1 sn sn 106K ápr 3 11:32 kimarad.fq.gz
-rw-rw-r-- 1 sn sn 453K ápr 3 11:32 marad.fq.gz
-rwxrwxr-x 1 sn sn 13K márc 19 12:04 minta_jelentes.Rmd
-rw-rw-r-- 1 sn sn 1,6M ápr 3 11:31 osszefoglalo.html
-rw-rw-r-- 1 sn sn 2,4M ápr 3 11:28 SRR1660259.fastq
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 9130 sequences (911661 bp)...
[M::mem_process_seqs] Processed 9130 reads in 0.323 CPU sec, 0.332 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem Ebola SRR1660259.fastq
[main] Real time: 0.479 sec; CPU: 0.344 sec
%%bash
cd gyak07
# samtools mpileup illesztes03_van_sorted.bam > illesztes03_van_sorted.pileup
samtools mpileup -f ebola1976.fa illesztes03_van_sorted.bam > illesztes03_van_sorted.pileup
tail illesztes03_van_sorted.pileup
AF086833.2 18924 T 4 ,,,, HHHF
AF086833.2 18925 A 4 ,,,, HFHH
AF086833.2 18926 A 4 ,,,, FDDD
AF086833.2 18927 A 4 ,,,, FDDD
AF086833.2 18928 A 4 ,,,, FDDD
AF086833.2 18929 A 4 ,,,, FDDB
AF086833.2 18930 T 4 ,,,, FDDD
AF086833.2 18931 A 4 ,,,, C@@@
AF086833.2 18932 A 4 ,,,, B@@@
AF086833.2 18933 A 4 ,$,$,$,$ @@@@
[mpileup] 1 samples in 1 input files
<mpileup> Set max per-file depth to 8000
Oszlopok jelentése: 1. a referencia szekvencia azonosítója 2. a szekvencia nukleotidjának pozíciója (1-ről induló sorszám) 3. a referencia nukleotid az adott pozíción 4. az adott pozíciót lefedő readek száma 5. az illesztett readek nukleotidja az adott pozícióban 6. az illesztett readek adott pozícióbeli nukleotidjának minősége
További információ: https://en.wikipedia.org/wiki/Pileup_format
library(Rsamtools)
bam.fajlom = 'illesztes03_van_sorted.bam'
countBam(bam.fajlom)
| space | start | end | width | file | records | nucleotides |
|---|---|---|---|---|---|---|
| NA | NA | NA | NA | illesztes03_van_sorted.bam | 9146 | 912260 |
idxstatsBam(bam.fajlom)
| seqnames | seqlength | mapped | unmapped |
|---|---|---|---|
| AF086833.2 | 18959 | 9146 | 0 |
# átlagos lefedettség
ref.szek.hossza = idxstatsBam(bam.fajlom)$seqlength
illesztett.nukleotidok.szama = countBam(bam.fajlom)$nucleotides
illesztett.nukleotidok.szama / ref.szek.hossza
olvasasi.parameterek = PileupParam(max_depth=50)
my.pileup = pileup(bam.fajlom, pileupParam=olvasasi.parameterek)
head(my.pileup)
| seqnames | pos | strand | nucleotide | count |
|---|---|---|---|---|
| AF086833.2 | 1 | + | C | 1 |
| AF086833.2 | 1 | - | C | 1 |
| AF086833.2 | 2 | + | G | 1 |
| AF086833.2 | 2 | - | G | 1 |
| AF086833.2 | 3 | + | G | 1 |
| AF086833.2 | 3 | - | G | 1 |
# http://ggplot2.org/
library(ggplot2)
ggplot(data=my.pileup, aes(x=pos, y=count)) + geom_point()
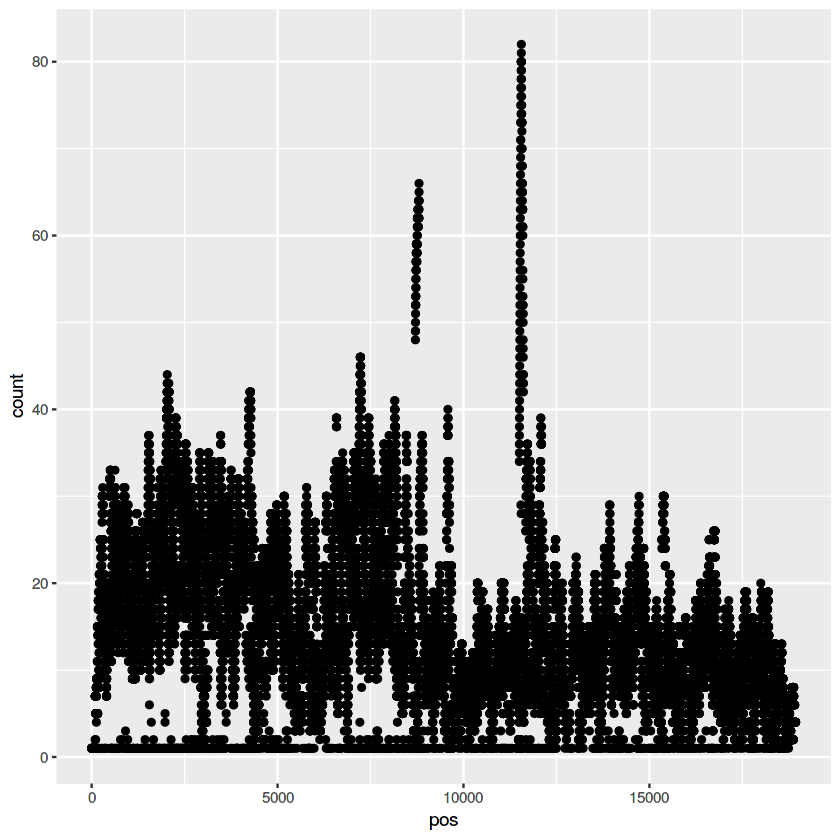
ggplot(data=my.pileup, aes(x=pos, y=count)) + geom_point() + theme_bw()
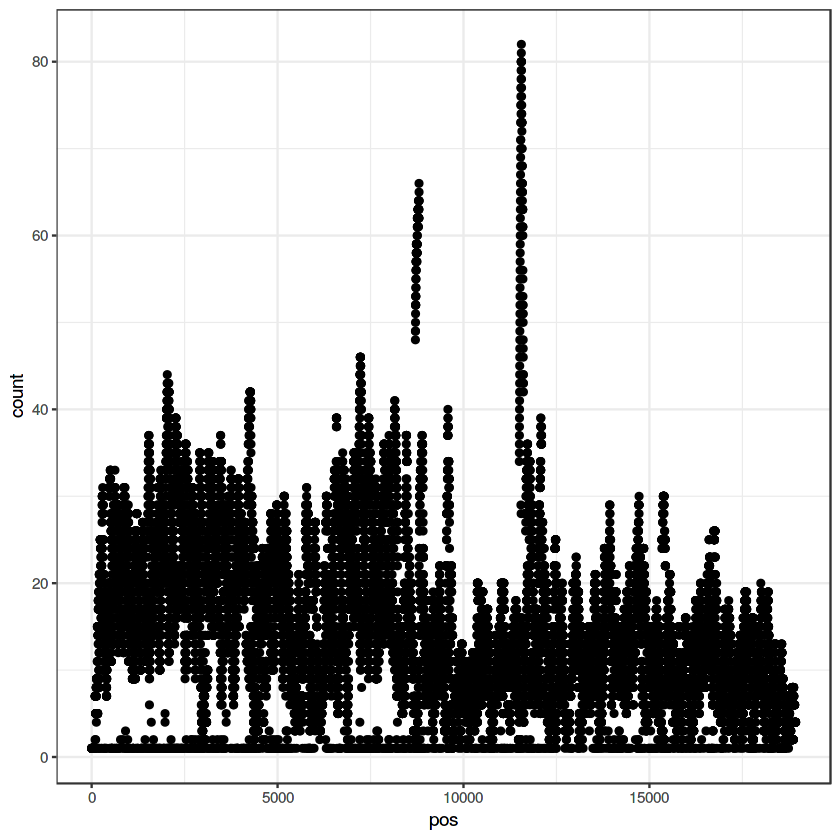
ggplot(data=my.pileup, aes(x=pos, y=count, color=strand)) + geom_point() + theme_bw()
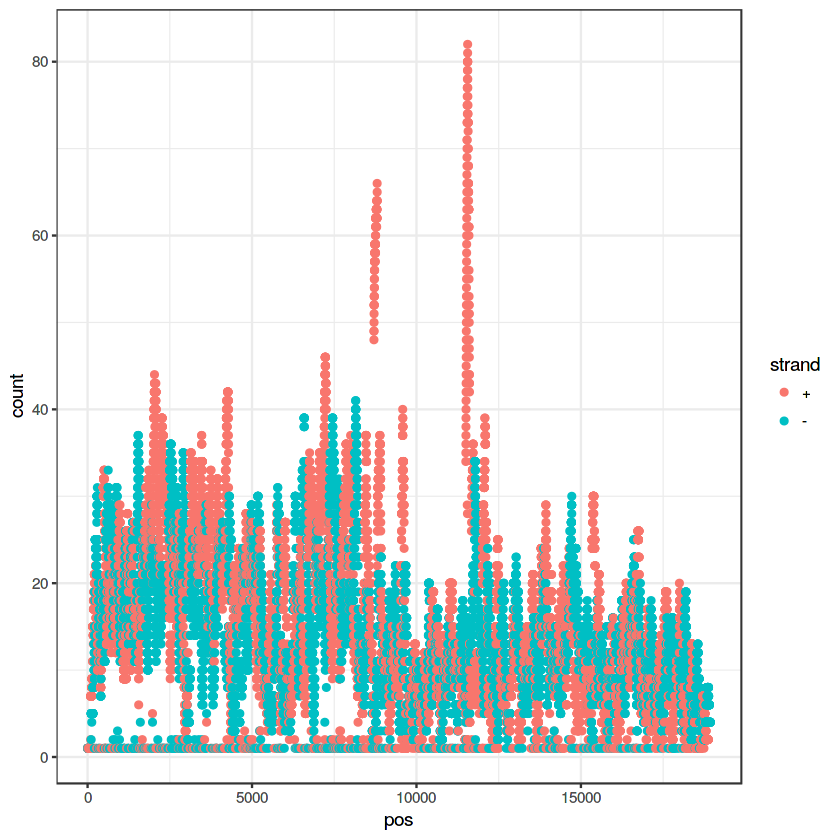
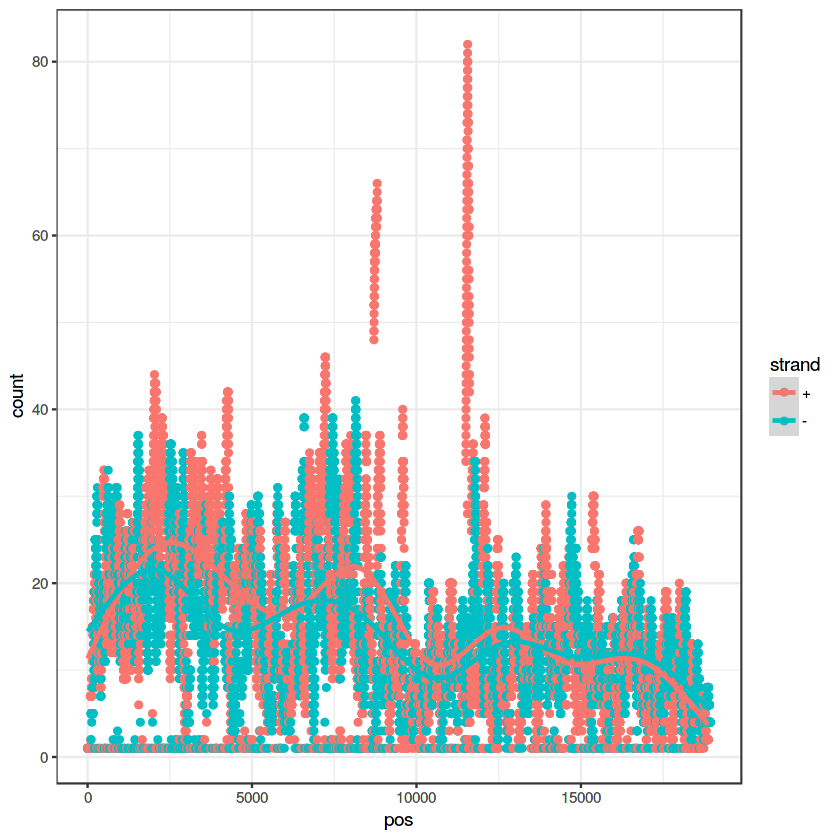
ggplot(data=my.pileup, aes(x=pos, y=count, color=strand)) + geom_point() + theme_bw() + geom_smooth()
geom_smooth() using method = 'gam'
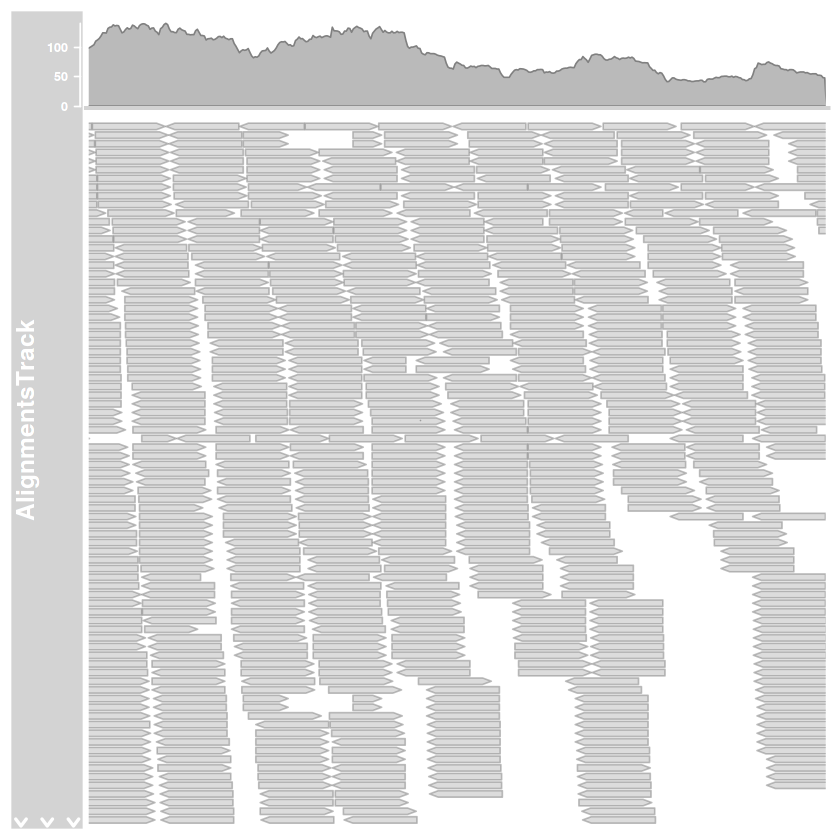
library(Gviz)
# https://www.bioconductor.org/packages/release/bioc/html/Gviz.html
# https://www.bioconductor.org/packages/release/bioc/vignettes/Gviz/inst/doc/Gviz.pdf
options(ucscChromosomeNames=FALSE)
# https://www.ncbi.nlm.nih.gov/nuccore/KM034562.1
# http://hgdownload.cse.ucsc.edu/downloads.html#ebola_virus
illesztes.track = AlignmentsTrack(bam.fajlom, start=2000, end=3000)
plotTracks(
list(illesztes.track),
type=c('coverage', 'pileup'),
chromosome='AF086833.2',
from=2000,
to=3000
)
Loading required package: grid
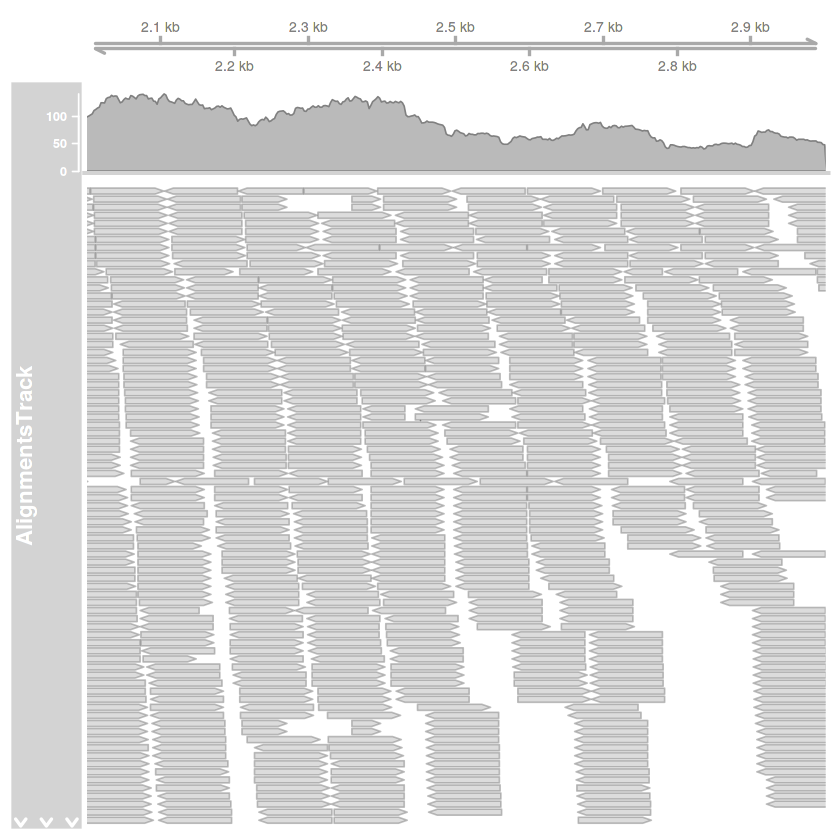
tengely.track = GenomeAxisTrack()
plotTracks(
list(tengely.track, illesztes.track),
type=c('coverage', 'pileup'),
chromosome='AF086833.2',
from=2000,
to=3000
)
library(rentrez)
library(seqinr)
ebola = entrez_fetch(db='nuccore', id='AF086833.2', rettype='fasta')
referencia = read.fasta(textConnection(ebola), as.string=TRUE, seqonly=TRUE)
referencia.szekvencia = referencia[[1]]
referencia.szekvencia = DNAStringSet(referencia.szekvencia)
names(referencia.szekvencia) = 'AF086833.2'
szekvencia.track = SequenceTrack(referencia.szekvencia)
plotTracks(
trackList = list(tengely.track, illesztes.track, szekvencia.track),
type = c('coverage', 'pileup'),
chromosome = 'AF086833.2',
from = 2000,
to = 3000
)
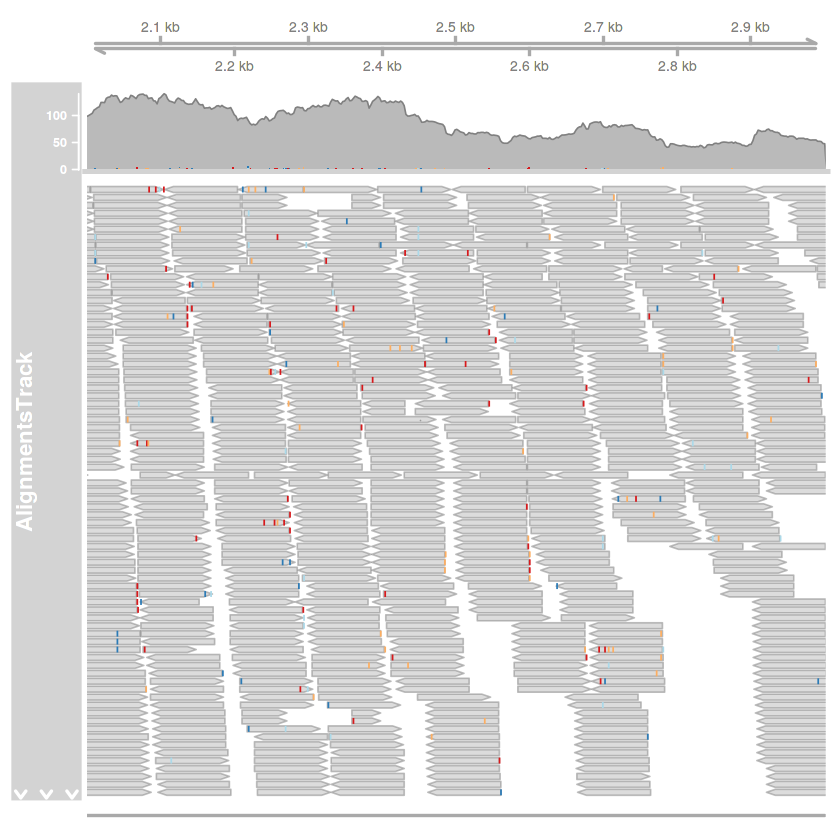
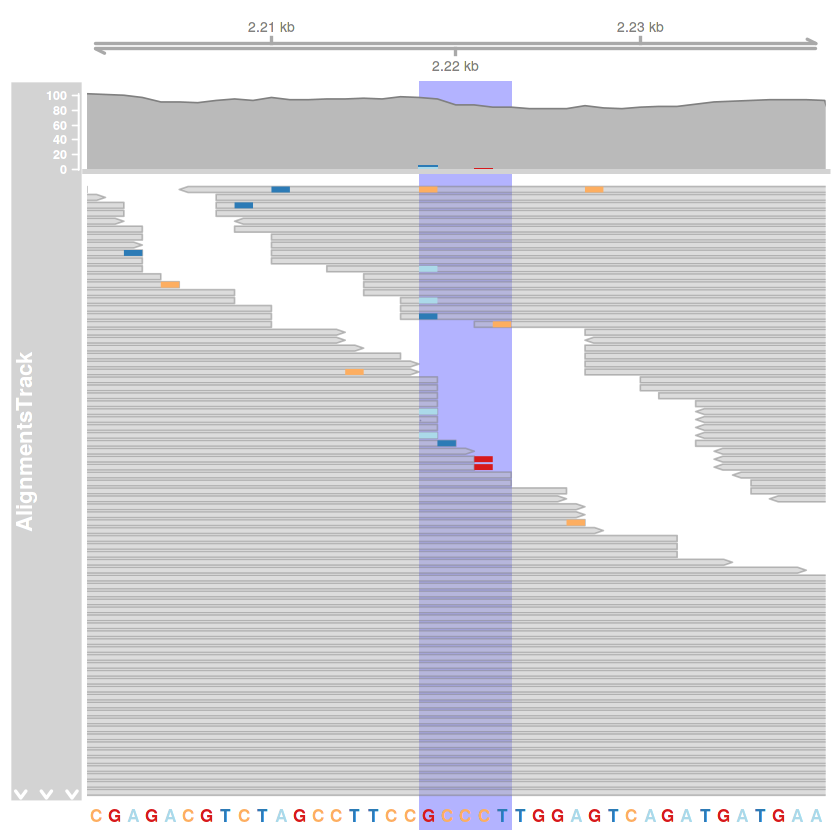
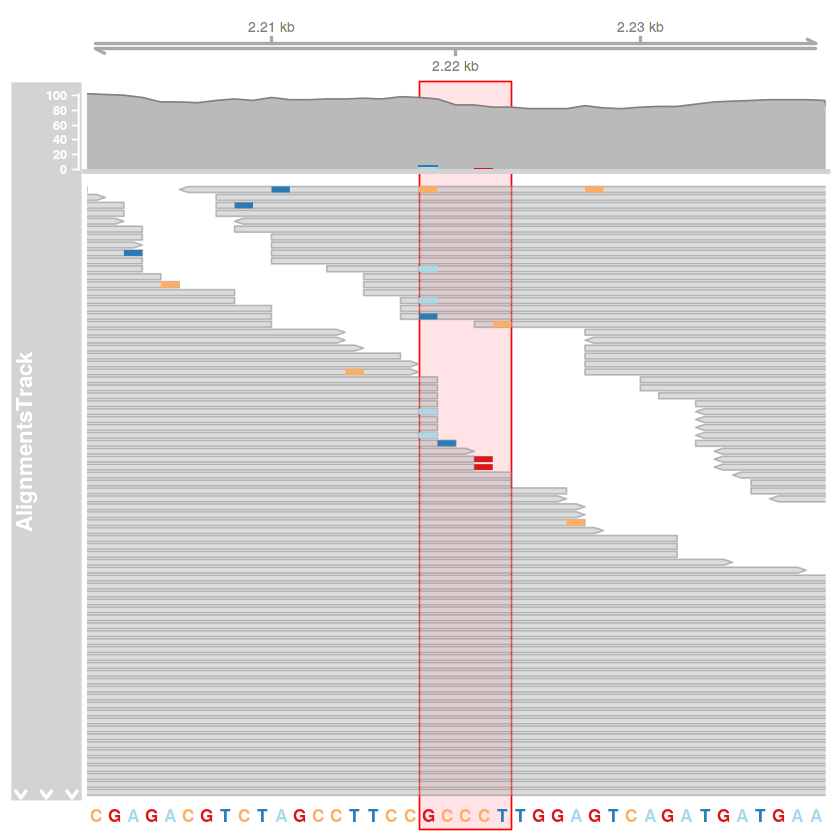
kiemeles = HighlightTrack(
trackList = list(illesztes.track, szekvencia.track),
chromosome = 'AF086833.2',
start = 2218,
end = 2222
)
plotTracks(
list(tengely.track, kiemeles),
type = c('coverage', 'pileup'),
chromosome = 'AF086833.2',
from = 2200,
to = 2240
)
displayPars(kiemeles)$fill = 'blue'
displayPars(kiemeles)$col = 'transparent'
displayPars(kiemeles)$alpha = 0.3
plotTracks(
list(tengely.track, kiemeles),
type = c('coverage', 'pileup'),
chromosome = 'AF086833.2',
from = 2200,
to = 2240
)