Variant Calling¶
- variant calling
- variant filtering
- variant annotation
Források: Genome Analysis in Ebola, viral-ngs
eboVir3.chrom.sizes-fájl alapján a szekvencia \(18957\)
nukleotidból áll.%%bash
mkdir gyak08
cd gyak08
wget http://hgdownload-test.cse.ucsc.edu/goldenPath/currentGenomes/Ebola_virus/bigZips/KM034562v1.fa.gz
gunzip KM034562v1.fa.gz
ls -lh
total 20K
-rw-rw-r-- 1 sn sn 19K Sep 30 2014 KM034562v1.fa
--2018-04-10 09:35:30-- http://hgdownload-test.cse.ucsc.edu/goldenPath/currentGenomes/Ebola_virus/bigZips/KM034562v1.fa.gz
Resolving hgdownload-test.cse.ucsc.edu (hgdownload-test.cse.ucsc.edu)... 132.249.245.79
Connecting to hgdownload-test.cse.ucsc.edu (hgdownload-test.cse.ucsc.edu)|132.249.245.79|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 6301 (6.2K) [application/x-gzip]
Saving to: ‘KM034562v1.fa.gz’
0K ...... 100% 30.6M=0s
2018-04-10 09:35:30 (30.6 MB/s) - ‘KM034562v1.fa.gz’ saved [6301/6301]
Az SRA SRR1553500 mintáját használjuk, amiben \(101\) nukleotid hosszúságú páros read-ek vannak. Ha átlagosan \(30\)-as lefedettséget biztosító readet szeretnénk csak letölteni az SRA-ról, akkor \(\cfrac{18957}{101}\times 30 \approx 5600\), vagyis kb. \(3000\) read-pár elég lehet.
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses, Nat Rev Genet. 2014 Feb;15(2):121-32. `doi: 10.1038/nrg3642 <https://www.nature.com/articles/nrg3642>`__.
%%bash
export PATH=$PATH:/usr/local/ncbi/sra-tools/bin
cd gyak08
fastq-dump -X 3000 --split-files SRR1553500
ls -lh
Read 3000 spots for SRR1553500
Written 3000 spots for SRR1553500
total 1.6M
-rw-rw-r-- 1 sn sn 19K Sep 30 2014 KM034562v1.fa
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_1.fastq
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_2.fastq
%%bash
export PATH=$PATH:/home/bioinfo/bwa
cd gyak08
bwa index -p ebolaRef KM034562v1.fa
ls -lh
total 1.7M
-rw-rw-r-- 1 sn sn 10 Apr 10 09:48 ebolaRef.amb
-rw-rw-r-- 1 sn sn 41 Apr 10 09:48 ebolaRef.ann
-rw-rw-r-- 1 sn sn 19K Apr 10 09:48 ebolaRef.bwt
-rw-rw-r-- 1 sn sn 4.7K Apr 10 09:48 ebolaRef.pac
-rw-rw-r-- 1 sn sn 9.4K Apr 10 09:48 ebolaRef.sa
-rw-rw-r-- 1 sn sn 19K Sep 30 2014 KM034562v1.fa
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_1.fastq
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_2.fastq
[bwa_index] Pack FASTA... 0.00 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 0.00 seconds elapse.
[bwa_index] Update BWT... 0.00 sec
[bwa_index] Pack forward-only FASTA... 0.00 sec
[bwa_index] Construct SA from BWT and Occ... 0.00 sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa index -p ebolaRef KM034562v1.fa
[main] Real time: 0.122 sec; CPU: 0.008 sec
%%bash
export PATH=$PATH:/home/bioinfo/bwa
cd gyak08
bwa mem ebolaRef SRR1553500_1.fastq SRR1553500_2.fastq | samtools view -Sb -F 4 | samtools sort > illesztes01.bam
samtools index illesztes01.bam
ls -lh
total 2.0M
-rw-rw-r-- 1 sn sn 10 Apr 10 09:48 ebolaRef.amb
-rw-rw-r-- 1 sn sn 41 Apr 10 09:48 ebolaRef.ann
-rw-rw-r-- 1 sn sn 19K Apr 10 09:48 ebolaRef.bwt
-rw-rw-r-- 1 sn sn 4.7K Apr 10 09:48 ebolaRef.pac
-rw-rw-r-- 1 sn sn 9.4K Apr 10 09:48 ebolaRef.sa
-rw-rw-r-- 1 sn sn 389K Apr 10 09:49 illesztes01.bam
-rw-rw-r-- 1 sn sn 152 Apr 10 09:49 illesztes01.bam.bai
-rw-rw-r-- 1 sn sn 19K Sep 30 2014 KM034562v1.fa
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_1.fastq
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_2.fastq
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 6000 sequences (606000 bp)...
[M::mem_pestat] # candidate unique pairs for (FF, FR, RF, RR): (234, 2511, 16, 206)
[M::mem_pestat] analyzing insert size distribution for orientation FF...
[M::mem_pestat] (25, 50, 75) percentile: (110, 187, 267)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 581)
[M::mem_pestat] mean and std.dev: (194.67, 117.50)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 738)
[M::mem_pestat] analyzing insert size distribution for orientation FR...
[M::mem_pestat] (25, 50, 75) percentile: (155, 260, 409)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 917)
[M::mem_pestat] mean and std.dev: (294.87, 177.15)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 1171)
[M::mem_pestat] analyzing insert size distribution for orientation RF...
[M::mem_pestat] (25, 50, 75) percentile: (65, 107, 189)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 437)
[M::mem_pestat] mean and std.dev: (114.27, 85.53)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 561)
[M::mem_pestat] analyzing insert size distribution for orientation RR...
[M::mem_pestat] (25, 50, 75) percentile: (103, 190, 307)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 715)
[M::mem_pestat] mean and std.dev: (213.14, 137.62)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 919)
[M::mem_pestat] skip orientation RF
[M::mem_process_seqs] Processed 6000 reads in 0.374 CPU sec, 0.373 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem ebolaRef SRR1553500_1.fastq SRR1553500_2.fastq
[main] Real time: 0.446 sec; CPU: 0.384 sec
%%bash
cd gyak08
samtools faidx KM034562v1.fa
freebayes -f KM034562v1.fa illesztes01.bam > sznipek_freebayes.vcf
ls -lh
total 2.1M
-rw-rw-r-- 1 sn sn 10 Apr 10 09:48 ebolaRef.amb
-rw-rw-r-- 1 sn sn 41 Apr 10 09:48 ebolaRef.ann
-rw-rw-r-- 1 sn sn 19K Apr 10 09:48 ebolaRef.bwt
-rw-rw-r-- 1 sn sn 4.7K Apr 10 09:48 ebolaRef.pac
-rw-rw-r-- 1 sn sn 9.4K Apr 10 09:48 ebolaRef.sa
-rw-rw-r-- 1 sn sn 389K Apr 10 09:49 illesztes01.bam
-rw-rw-r-- 1 sn sn 152 Apr 10 09:49 illesztes01.bam.bai
-rw-rw-r-- 1 sn sn 19K Sep 30 2014 KM034562v1.fa
-rw-rw-r-- 1 sn sn 26 Apr 10 10:02 KM034562v1.fa.fai
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_1.fastq
-rw-rw-r-- 1 sn sn 787K Apr 10 09:47 SRR1553500_2.fastq
-rw-rw-r-- 1 sn sn 24K Apr 10 10:02 sznipek_freebayes.vcf
## TERMINÁL ##
cd gyak08
less sznipek_freebayes.vcf
Variant Call Format (VCF) fájl¶
What is a VCF and how should I interpret it?
Header¶
##fileformat=VCFv4.2
##fileDate=20180405
##source=freeBayes v1.1.0-60-gc15b070
##reference=KM034562v1.fa
##contig=<ID=KM034562v1,length=18957>
##phasing=none
##commandline="freebayes -f KM034562v1.fa illesztes01.bam"
##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of samples with data">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total read depth at the locus">
##INFO=<ID=DPB,Number=1,Type=Float,Description="Total read depth per bp at the locus; bases in reads overlapping / bases in haplotype">
##INFO=<ID=AC,Number=A,Type=Integer,Description="Total number of alternate alleles in called genotypes">
##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes">
##INFO=<ID=AF,Number=A,Type=Float,Description="Estimated allele frequency in the range (0,1]">
##INFO=<ID=RO,Number=1,Type=Integer,Description="Count of full observations of the reference haplotype.">
##INFO=<ID=AO,Number=A,Type=Integer,Description="Count of full observations of this alternate haplotype.">
##INFO=<ID=PRO,Number=1,Type=Float,Description="Reference allele observation count, with partial observations recorded fractionally">
##INFO=<ID=PAO,Number=A,Type=Float,Description="Alternate allele observations, with partial observations recorded fractionally">
##INFO=<ID=QR,Number=1,Type=Integer,Description="Reference allele quality sum in phred">
##INFO=<ID=QA,Number=A,Type=Integer,Description="Alternate allele quality sum in phred">
##INFO=<ID=PQR,Number=1,Type=Float,Description="Reference allele quality sum in phred for partial observations">
##INFO=<ID=PQA,Number=A,Type=Float,Description="Alternate allele quality sum in phred for partial observations">
##INFO=<ID=SRF,Number=1,Type=Integer,Description="Number of reference observations on the forward strand">
##INFO=<ID=SRR,Number=1,Type=Integer,Description="Number of reference observations on the reverse strand">
##INFO=<ID=SAF,Number=A,Type=Integer,Description="Number of alternate observations on the forward strand">
##INFO=<ID=SAR,Number=A,Type=Integer,Description="Number of alternate observations on the reverse strand">
##INFO=<ID=SRP,Number=1,Type=Float,Description="Strand balance probability for the reference allele: Phred-scaled upper-bounds estimate of the probability of observing the deviation between SRF and SRR given E(SRF/SRR) ~ 0.5, derived using Hoeffding's inequality">
##INFO=<ID=SAP,Number=A,Type=Float,Description="Strand balance probability for the alternate allele: Phred-scaled upper-bounds estimate of the probability of observing the deviation between SAF and SAR given E(SAF/SAR) ~ 0.5, derived using Hoeffding's inequality">
##INFO=<ID=AB,Number=A,Type=Float,Description="Allele balance at heterozygous sites: a number between 0 and 1 representing the ratio of reads showing the reference allele to all reads, considering only reads from individuals called as heterozygous">
##INFO=<ID=ABP,Number=A,Type=Float,Description="Allele balance probability at heterozygous sites: Phred-scaled upper-bounds estimate of the probability of observing the deviation between ABR and ABA given E(ABR/ABA) ~ 0.5, derived using Hoeffding's inequality">
##INFO=<ID=RUN,Number=A,Type=Integer,Description="Run length: the number of consecutive repeats of the alternate allele in the reference genome">
##INFO=<ID=RPP,Number=A,Type=Float,Description="Read Placement Probability: Phred-scaled upper-bounds estimate of the probability of observing the deviation between RPL and RPR given E(RPL/RPR) ~ 0.5, derived using Hoeffding's inequality">
##INFO=<ID=RPPR,Number=1,Type=Float,Description="Read Placement Probability for reference observations: Phred-scaled upper-bounds estimate of the probability of observing the deviation between RPL and RPR given E(RPL/RPR) ~ 0.5, derived using Hoeffding's inequality">
##INFO=<ID=RPL,Number=A,Type=Float,Description="Reads Placed Left: number of reads supporting the alternate balanced to the left (5') of the alternate allele">
##INFO=<ID=RPR,Number=A,Type=Float,Description="Reads Placed Right: number of reads supporting the alternate balanced to the right (3') of the alternate allele">
##INFO=<ID=EPP,Number=A,Type=Float,Description="End Placement Probability: Phred-scaled upper-bounds estimate of the probability of observing the deviation between EL and ER given E(EL/ER) ~ 0.5, derived using Hoeffding's inequality">
##INFO=<ID=EPPR,Number=1,Type=Float,Description="End Placement Probability for reference observations: Phred-scaled upper-bounds estimate of the probability of observing the deviation between EL and ER given E(EL/ER) ~ 0.5, derived using Hoeffding's inequality">
##INFO=<ID=DPRA,Number=A,Type=Float,Description="Alternate allele depth ratio. Ratio between depth in samples with each called alternate allele and those without.">
##INFO=<ID=ODDS,Number=1,Type=Float,Description="The log odds ratio of the best genotype combination to the second-best.">
##INFO=<ID=GTI,Number=1,Type=Integer,Description="Number of genotyping iterations required to reach convergence or bailout.">
##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex.">
##INFO=<ID=CIGAR,Number=A,Type=String,Description="The extended CIGAR representation of each alternate allele, with the exception that '=' is replaced by 'M' to ease VCF parsing. Note that INDEL alleles do not have the first matched base (which is provided by default, per the spec) referred to by the CIGAR.">
##INFO=<ID=NUMALT,Number=1,Type=Integer,Description="Number of unique non-reference alleles in called genotypes at this position.">
##INFO=<ID=MEANALT,Number=A,Type=Float,Description="Mean number of unique non-reference allele observations per sample with the corresponding alternate alleles.">
##INFO=<ID=LEN,Number=A,Type=Integer,Description="allele length">
##INFO=<ID=MQM,Number=A,Type=Float,Description="Mean mapping quality of observed alternate alleles">
##INFO=<ID=MQMR,Number=1,Type=Float,Description="Mean mapping quality of observed reference alleles">
##INFO=<ID=PAIRED,Number=A,Type=Float,Description="Proportion of observed alternate alleles which are supported by properly paired read fragments">
##INFO=<ID=PAIREDR,Number=1,Type=Float,Description="Proportion of observed reference alleles which are supported by properly paired read fragments">
##INFO=<ID=MIN_DP,Number=1,Type=Integer,Description="Minimum depth in gVCF output block.">
##INFO=<ID=END,Number=1,Type=Integer,Description="Last position (inclusive) in gVCF output record.">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Genotype Quality, the Phred-scaled marginal (or unconditional) probability of the called genotype">
##FORMAT=<ID=GL,Number=G,Type=Float,Description="Genotype Likelihood, log10-scaled likelihoods of the data given the called genotype for each possible genotype generated from the reference and alternate alleles given the sample ploidy">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth">
##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Number of observation for each allele">
##FORMAT=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count">
##FORMAT=<ID=QR,Number=1,Type=Integer,Description="Sum of quality of the reference observations">
##FORMAT=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observation count">
##FORMAT=<ID=QA,Number=A,Type=Integer,Description="Sum of quality of the alternate observations">
##FORMAT=<ID=MIN_DP,Number=1,Type=Integer,Description="Minimum depth in gVCF output block.">
Records¶
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT unknown
KM034562v1 800 . C T 874.877 . AB=0;ABP=0;AC=2;AF=1;AN=2;AO=28;CIGAR=1X;DP=28;DPB=28;DPRA=0;EPP=3.32051;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=43.4214;PAIRED=1;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1001;QR=0;RO=0;RPL=12;RPP=4.25114;RPPR=0;RPR=16;RUN=1;SAF=11;SAP=5.80219;SAR=17;SRF=0;SRP=0;SRR=0;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 1/1:28:0,28:0:0:28:1001:-90.3858,-8.42884,0
KM034562v1 8928 . A C 909.973 . AB=0;ABP=0;AC=2;AF=1;AN=2;AO=28;CIGAR=1X;DP=28;DPB=28;DPRA=0;EPP=7.97367;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=43.4214;PAIRED=0.964286;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1044;QR=0;RO=0;RPL=13;RPP=3.32051;RPPR=0;RPR=15;RUN=1;SAF=17;SAP=5.80219;SAR=11;SRF=0;SRP=0;SRR=0;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 1/1:28:0,28:0:0:28:1044:-94.2605,-8.42884,0
KM034562v1 10218 . G A 865.247 . AB=0;ABP=0;AC=2;AF=1;AN=2;AO=28;CIGAR=1X;DP=28;DPB=28;DPRA=0;EPP=3.32051;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=43.4214;PAIRED=1;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=989;QR=0;RO=0;RPL=14;RPP=3.0103;RPPR=0;RPR=14;RUN=1;SAF=17;SAP=5.80219;SAR=11;SRF=0;SRP=0;SRR=0;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 1/1:28:0,28:0:0:28:989:-89.3026,-8.42884,0
KM034562v1 11811 . T C 919.423 . AB=0;ABP=0;AC=2;AF=1;AN=2;AO=27;CIGAR=1X;DP=27;DPB=27;DPRA=0;EPP=3.09072;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=42.0351;PAIRED=1;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1050;QR=0;RO=0;RPL=14;RPP=3.09072;RPPR=0;RPR=13;RUN=1;SAF=17;SAP=6.95112;SAR=10;SRF=0;SRP=0;SRR=0;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 1/1:27:0,27:0:0:27:1050:-94.795,-8.12781,0
KM034562v1 15599 . G A 1672.84 . AB=0;ABP=0;AC=2;AF=1;AN=2;AO=50;CIGAR=1X;DP=50;DPB=50;DPRA=0;EPP=3.70517;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=73.9199;PAIRED=0.98;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1891;QR=0;RO=0;RPL=28;RPP=4.57376;RPPR=0;RPR=22;RUN=1;SAF=27;SAP=3.70517;SAR=23;SRF=0;SRP=0;SRR=0;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 1/1:50:0,50:0:0:50:1891:-170.423,-15.0515,0
KM034562v1 15963 . G A 930.435 . AB=0;ABP=0;AC=2;AF=1;AN=2;AO=27;CIGAR=1X;DP=27;DPB=27;DPRA=0;EPP=6.95112;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=42.0351;PAIRED=1;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1070;QR=0;RO=0;RPL=18;RPP=9.52472;RPPR=0;RPR=9;RUN=1;SAF=15;SAP=3.73412;SAR=12;SRF=0;SRP=0;SRR=0;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 1/1:27:0,27:0:0:27:1070:-96.5897,-8.12781,0
KM034562v1 17142 . T C 1423.48 . AB=0;ABP=0;AC=2;AF=1;AN=2;AO=42;CIGAR=1X;DP=42;DPB=42;DPRA=0;EPP=6.31921;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60;MQMR=0;NS=1;NUMALT=1;ODDS=62.8295;PAIRED=1;PAIREDR=0;PAO=0;PQA=0;PQR=0;PRO=0;QA=1614;QR=0;RO=0;RPL=22;RPP=3.21711;RPPR=0;RPR=20;RUN=1;SAF=19;SAP=3.83753;SAR=23;SRF=0;SRP=0;SRR=0;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 1/1:42:0,42:0:0:42:1614:-145.511,-12.6433,0
Az első record:¶
| Mező | Érték | Mező |
|---|---|---|
| CHROM | KM034562v1 | CHROM |
| POS | 800 | POS |
| ID | . | ID |
| REF | C | REF |
| ALT | T | ALT |
| QUAL | 874.877 | QUAL |
| FILTER | . | FILTER |
| INFO | AB=0;ABP=0;AC=2;AF=1;AN=2;AO=28;CIGAR=1X;DP=28;DPB=28; DPRA=0;EPP=3.32051;EPPR=0;GTI=0;LEN=1;MEANALT=1;MQM=60 ;MQMR=0;NS=1;NUMALT=1;ODDS=43.4214;PAIRED=1;PAIREDR=0; PAO=0;PQA=0;PQR=0;PRO=0;QA=1001;QR=0;RO=0;RPL=12;RPP=4 .25114;RPPR=0;RPR=16;RUN=1;SAF=11;SAP=5.80219;SAR=17;S RF=0;SRP=0;SRR=0;TYPE=snp | INFO |
| FORMAT | GT:DP:AD:RO:QR:AO:QA:GL | FORMAT |
| unknown | 1/1:28:0,28:0:0:28:1001:-90.3858,-8.42884,0 | unknown |
INFO:¶
| Mező | Érték |
|---|---|
| AB | 0 |
| ABP | 0 |
| AC | 2 |
| AF | 1 |
| AN | 2 |
| AO | 28 |
| CIGAR | 1X |
| DP | 28 |
| DPB | 28 |
| DPRA | 0 |
| EPP | 3.32051 |
| EPPR | 0 |
| GTI | 0 |
| LEN | 1 |
| MEANALT | 1 |
| MQM | 60 |
| MQMR | 0 |
| NS | 1 |
| NUMALT | 1 |
| ODDS | 43.4214 |
| PAIRED | 1 |
| PAIREDR | 0 |
| PAO | 0 |
| PQA | 0 |
| PQR | 0 |
| PRO | 0 |
| QA | 1001 |
| QR | 0 |
| RO | 0 |
| RPL | 12 |
| RPP | 4.25114 |
| RPPR | 0 |
| RPR | 16 |
| RUN | 1 |
| SAF | 11 |
| SAP | 5.80219 |
| SAR | 17 |
| SRF | 0 |
| SRP | 0 |
| SRR | 0 |
| TYPE | snp |
FORMAT:¶
| FORMAT | unknown |
|---|---|
| GT | 1/1 |
| DP | 28 |
| AD | 0,28 |
| RO | 0 |
| QR | 0 |
| AO | 28 |
| QA | 1001 |
| GL | -90.3858,-8.42884,0 |
Genomic VCF (gVCF) fájl¶
https://software.broadinstitute.org/gatk/documentation/article.php?id=4017

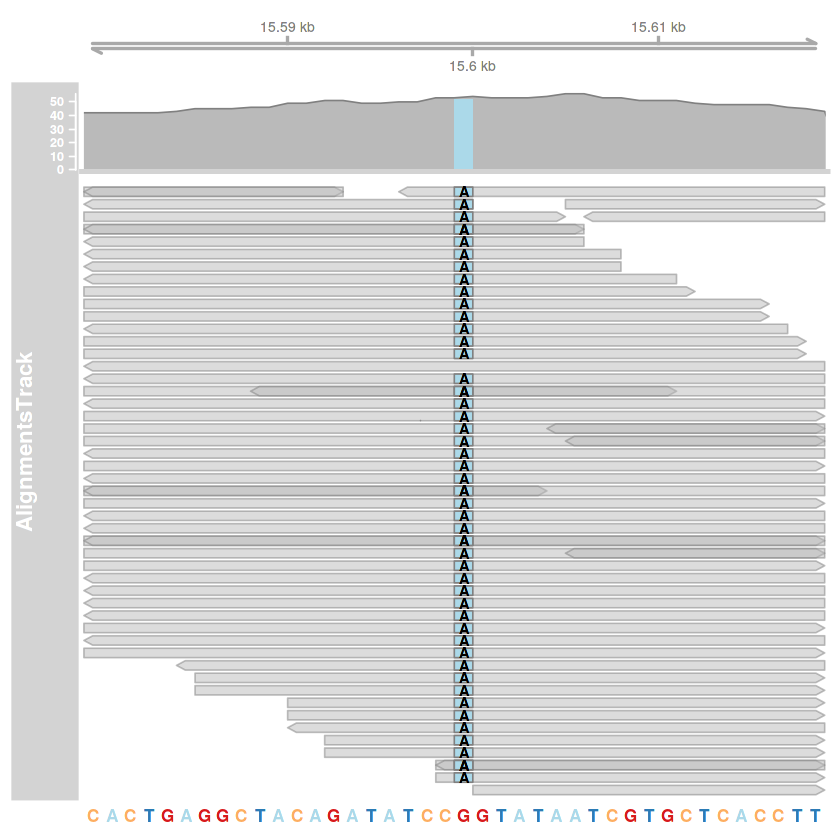
title
# R
library(Gviz)
library(seqinr)
library(Biostrings)
options(ucscChromosomeNames=FALSE)
setwd('gyak08')
# a 15599. pozíció ábrázolása
poz = 15599
kezdet = poz - 20
veg = poz + 20
bam.fajlom = 'illesztes01.bam'
illesztes.track = AlignmentsTrack(bam.fajlom, start=kezdet, end=veg)
tengely.track = GenomeAxisTrack()
referencia = read.fasta('KM034562v1.fa', as.string=TRUE, seqonly=TRUE)
referencia.szekvencia = referencia[[1]]
referencia.szekvencia = DNAStringSet(referencia.szekvencia)
names(referencia.szekvencia) = 'KM034562v1'
szekvencia.track = SequenceTrack(referencia.szekvencia)
plotTracks(
list(tengely.track, illesztes.track, szekvencia.track),
type=c('coverage', 'pileup'),
chromosome='KM034562v1', #!!!!!!!
from=kezdet,
to=veg
)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, cbind, colMeans, colnames,
colSums, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, lengths, Map, mapply, match,
mget, order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rowMeans, rownames, rowSums, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which, which.max, which.min
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:base’:
expand.grid
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: grid
Loading required package: XVector
Attaching package: 'Biostrings'
The following object is masked from 'package:seqinr':
translate
The following object is masked from 'package:base':
strsplit
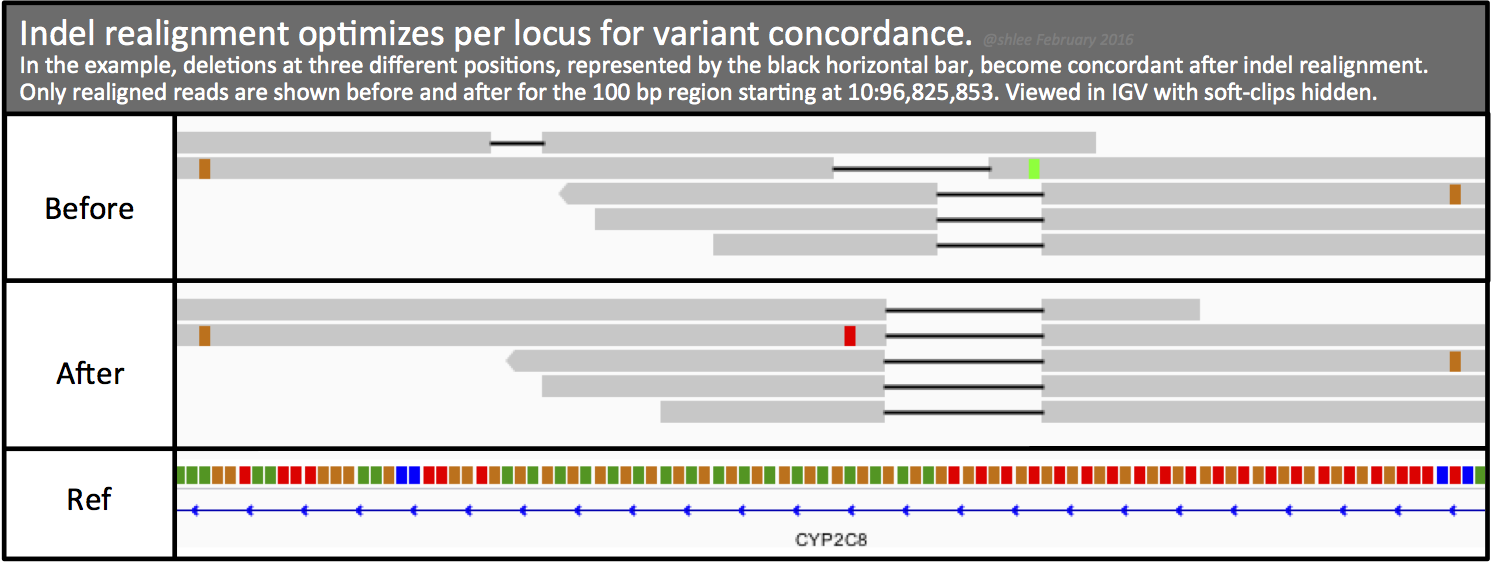
GATK Best Practice¶


https://qcb.ucla.edu/wp-content/uploads/sites/14/2016/03/GATKwr12-2-Marking_duplicates.pdf
%%bash
cd gyak08
# duplum readek eltávolítása
# https://broadinstitute.github.io/picard/index.html
PIKARD='/home/bioinfo/tools/picard/build/libs/picard.jar'
java -jar $PIKARD MarkDuplicates \
INPUT=illesztes01.bam \
OUTPUT=illesztes01_deduplikalt.bam \
METRICS_FILE=duplum_metrics.txt \
VALIDATION_STRINGENCY=LENIENT \
ASSUME_SORTED=true \
REMOVE_DUPLICATES=true
# a GATK-hoz kellenek:
# minden readhez rendelnünk kell egy új csoportazonosítót
java -jar $PIKARD AddOrReplaceReadGroups INPUT=illesztes01_deduplikalt.bam OUTPUT=illesztes01_deduplikalt_grp.bam RGID=4 RGLB=lib1 RGPL=illumina RGPU=unit1 RGSM=20
# érdekes módon, ha az előző sortörést alkalmazzuk, akkor nem fut le :(
# a referencia fasta-fájlhoz létre kell hozni egy dictionary-t
java -jar $PIKARD CreateSequenceDictionary R=KM034562v1.fa O=KM034562v1.dict
# samtools dict KM034562v1.fa > KM034562v1.dict
10:40:23.070 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home/bioinfo/tools/picard/build/libs/picard.jar!/com/intel/gkl/native/libgkl_compression.so
[Tue Apr 10 10:40:23 CEST 2018] MarkDuplicates INPUT=[illesztes01.bam] OUTPUT=illesztes01_deduplikalt.bam METRICS_FILE=duplum_metrics.txt REMOVE_DUPLICATES=true ASSUME_SORTED=true VALIDATION_STRINGENCY=LENIENT MAX_SEQUENCES_FOR_DISK_READ_ENDS_MAP=50000 MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=8000 SORTING_COLLECTION_SIZE_RATIO=0.25 TAG_DUPLICATE_SET_MEMBERS=false REMOVE_SEQUENCING_DUPLICATES=false TAGGING_POLICY=DontTag CLEAR_DT=true ADD_PG_TAG_TO_READS=true DUPLICATE_SCORING_STRATEGY=SUM_OF_BASE_QUALITIES PROGRAM_RECORD_ID=MarkDuplicates PROGRAM_GROUP_NAME=MarkDuplicates READ_NAME_REGEX=<optimized capture of last three ':' separated fields as numeric values> OPTICAL_DUPLICATE_PIXEL_DISTANCE=100 MAX_OPTICAL_DUPLICATE_SET_SIZE=300000 VERBOSITY=INFO QUIET=false COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
[Tue Apr 10 10:40:23 CEST 2018] Executing as sn@sn-OptiPlex-3010 on Linux 4.13.0-38-generic amd64; OpenJDK 64-Bit Server VM 1.8.0_162-8u162-b12-0ubuntu0.16.04.2-b12; Deflater: Intel; Inflater: Intel; Provider GCS is not available; Picard version: 2.18.2-SNAPSHOT
INFO 2018-04-10 10:40:23 MarkDuplicates Start of doWork freeMemory: 151835592; totalMemory: 157286400; maxMemory: 1836580864
INFO 2018-04-10 10:40:23 MarkDuplicates Reading input file and constructing read end information.
INFO 2018-04-10 10:40:23 MarkDuplicates Will retain up to 6654278 data points before spilling to disk.
WARNING 2018-04-10 10:40:23 AbstractOpticalDuplicateFinderCommandLineProgram A field field parsed out of a read name was expected to contain an integer and did not. Read name: SRR1553500.92. Cause: String 'SRR1553500.92' did not start with a parsable number.
INFO 2018-04-10 10:40:23 MarkDuplicates Read 6304 records. 0 pairs never matched.
INFO 2018-04-10 10:40:23 MarkDuplicates After buildSortedReadEndLists freeMemory: 191005096; totalMemory: 250085376; maxMemory: 1836580864
INFO 2018-04-10 10:40:23 MarkDuplicates Will retain up to 57393152 duplicate indices before spilling to disk.
INFO 2018-04-10 10:40:23 MarkDuplicates Traversing read pair information and detecting duplicates.
INFO 2018-04-10 10:40:23 MarkDuplicates Traversing fragment information and detecting duplicates.
INFO 2018-04-10 10:40:23 MarkDuplicates Sorting list of duplicate records.
INFO 2018-04-10 10:40:23 MarkDuplicates After generateDuplicateIndexes freeMemory: 249788208; totalMemory: 714080256; maxMemory: 1836580864
INFO 2018-04-10 10:40:23 MarkDuplicates Marking 2 records as duplicates.
INFO 2018-04-10 10:40:23 MarkDuplicates Found 0 optical duplicate clusters.
INFO 2018-04-10 10:40:23 MarkDuplicates Reads are assumed to be ordered by: coordinate
INFO 2018-04-10 10:40:23 MarkDuplicates Before output close freeMemory: 703788928; totalMemory: 709361664; maxMemory: 1836580864
INFO 2018-04-10 10:40:23 MarkDuplicates After output close freeMemory: 708583000; totalMemory: 714080256; maxMemory: 1836580864
[Tue Apr 10 10:40:23 CEST 2018] picard.sam.markduplicates.MarkDuplicates done. Elapsed time: 0.01 minutes.
Runtime.totalMemory()=714080256
10:40:24.453 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home/bioinfo/tools/picard/build/libs/picard.jar!/com/intel/gkl/native/libgkl_compression.so
[Tue Apr 10 10:40:24 CEST 2018] AddOrReplaceReadGroups INPUT=illesztes01_deduplikalt.bam OUTPUT=illesztes01_deduplikalt_grp.bam RGID=4 RGLB=lib1 RGPL=illumina RGPU=unit1 RGSM=20 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
[Tue Apr 10 10:40:24 CEST 2018] Executing as sn@sn-OptiPlex-3010 on Linux 4.13.0-38-generic amd64; OpenJDK 64-Bit Server VM 1.8.0_162-8u162-b12-0ubuntu0.16.04.2-b12; Deflater: Intel; Inflater: Intel; Provider GCS is not available; Picard version: 2.18.2-SNAPSHOT
INFO 2018-04-10 10:40:24 AddOrReplaceReadGroups Created read-group ID=4 PL=illumina LB=lib1 SM=20
[Tue Apr 10 10:40:24 CEST 2018] picard.sam.AddOrReplaceReadGroups done. Elapsed time: 0.00 minutes.
Runtime.totalMemory()=124780544
10:40:25.237 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home/bioinfo/tools/picard/build/libs/picard.jar!/com/intel/gkl/native/libgkl_compression.so
[Tue Apr 10 10:40:25 CEST 2018] CreateSequenceDictionary OUTPUT=KM034562v1.dict REFERENCE=KM034562v1.fa TRUNCATE_NAMES_AT_WHITESPACE=true NUM_SEQUENCES=2147483647 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
[Tue Apr 10 10:40:25 CEST 2018] Executing as sn@sn-OptiPlex-3010 on Linux 4.13.0-38-generic amd64; OpenJDK 64-Bit Server VM 1.8.0_162-8u162-b12-0ubuntu0.16.04.2-b12; Deflater: Intel; Inflater: Intel; Provider GCS is not available; Picard version: 2.18.2-SNAPSHOT
[Tue Apr 10 10:40:25 CEST 2018] picard.sam.CreateSequenceDictionary done. Elapsed time: 0.00 minutes.
Runtime.totalMemory()=124780544
%%bash
cd gyak08
# https://fathom.info/mirador/ebola/datarelease/
# wget https://raw.githubusercontent.com/mirador/ebola-data/master/sources/vcf/SNP-2014.vcf
wget https://github.com/mirador/ebola-data/releases/download/1.3/ebola-raw.zip
unzip ebola-raw.zip
ls -lh ebola-raw/vcf
Archive: ebola-raw.zip
creating: ebola-raw/
creating: ebola-raw/csv/
inflating: ebola-raw/csv/CaseNotification_schieffelin.csv
inflating: ebola-raw/csv/DemographicsFromSim_schieffelin.csv
inflating: ebola-raw/csv/FinalPiccoloData_schieffelin-FinalSummary1.csv
inflating: ebola-raw/csv/MasterDataListandEBOVResults.csv
creating: ebola-raw/vcf/
inflating: ebola-raw/vcf/clusters.tsv
inflating: ebola-raw/vcf/iSNV-all.vcf
inflating: ebola-raw/vcf/SNP-2014.vcf
creating: ebola-raw/xls/
inflating: ebola-raw/xls/CaseNotification_schieffelin.xlsx
creating: __MACOSX/
creating: __MACOSX/ebola-raw/
creating: __MACOSX/ebola-raw/xls/
inflating: __MACOSX/ebola-raw/xls/._CaseNotification_schieffelin.xlsx
inflating: ebola-raw/xls/DemographicsFromSim_schieffelin.xlsx
inflating: ebola-raw/xls/FinalPiccoloData_schieffelin.xlsx
inflating: ebola-raw/xls/info.docx
inflating: __MACOSX/ebola-raw/xls/._info.docx
inflating: ebola-raw/xls/MasterDataListandEBOVResults.xlsx
inflating: __MACOSX/ebola-raw/xls/._MasterDataListandEBOVResults.xlsx
total 192K
-rw-r--r-- 1 sn sn 2.2K Jan 15 2015 clusters.tsv
-rw-r--r-- 1 sn sn 166K Jan 15 2015 iSNV-all.vcf
-rw-r--r-- 1 sn sn 18K Jan 15 2015 SNP-2014.vcf
--2018-04-10 10:49:11-- https://github.com/mirador/ebola-data/releases/download/1.3/ebola-raw.zip
Resolving github.com (github.com)... 192.30.253.112, 192.30.253.113
Connecting to github.com (github.com)|192.30.253.112|:443... connected.
HTTP request sent, awaiting response... 302 Found
Location: https://github-production-release-asset-2e65be.s3.amazonaws.com/27739369/84259d3e-a558-11e4-9e26-219a9a054b51?X-Amz-Algorithm=AWS4-HMAC-SHA256&X-Amz-Credential=AKIAIWNJYAX4CSVEH53A%2F20180410%2Fus-east-1%2Fs3%2Faws4_request&X-Amz-Date=20180410T084830Z&X-Amz-Expires=300&X-Amz-Signature=9e8aa99c39e5f290566ff523e19dff28e5f3a7b39c737954049dbe2de7ec2e0c&X-Amz-SignedHeaders=host&actor_id=0&response-content-disposition=attachment%3B%20filename%3Debola-raw.zip&response-content-type=application%2Foctet-stream [following]
--2018-04-10 10:49:12-- https://github-production-release-asset-2e65be.s3.amazonaws.com/27739369/84259d3e-a558-11e4-9e26-219a9a054b51?X-Amz-Algorithm=AWS4-HMAC-SHA256&X-Amz-Credential=AKIAIWNJYAX4CSVEH53A%2F20180410%2Fus-east-1%2Fs3%2Faws4_request&X-Amz-Date=20180410T084830Z&X-Amz-Expires=300&X-Amz-Signature=9e8aa99c39e5f290566ff523e19dff28e5f3a7b39c737954049dbe2de7ec2e0c&X-Amz-SignedHeaders=host&actor_id=0&response-content-disposition=attachment%3B%20filename%3Debola-raw.zip&response-content-type=application%2Foctet-stream
Resolving github-production-release-asset-2e65be.s3.amazonaws.com (github-production-release-asset-2e65be.s3.amazonaws.com)... 52.216.97.27
Connecting to github-production-release-asset-2e65be.s3.amazonaws.com (github-production-release-asset-2e65be.s3.amazonaws.com)|52.216.97.27|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 534670 (522K) [application/octet-stream]
Saving to: ‘ebola-raw.zip’
0K .......... .......... .......... .......... .......... 9% 230K 2s
50K .......... .......... .......... .......... .......... 19% 228K 2s
100K .......... .......... .......... .......... .......... 28% 1.40M 1s
150K .......... .......... .......... .......... .......... 38% 626K 1s
200K .......... .......... .......... .......... .......... 47% 515K 1s
250K .......... .......... .......... .......... .......... 57% 3.08M 0s
300K .......... .......... .......... .......... .......... 67% 10.3M 0s
350K .......... .......... .......... .......... .......... 76% 518K 0s
400K .......... .......... .......... .......... .......... 86% 3.80M 0s
450K .......... .......... .......... .......... .......... 95% 11.4M 0s
500K .......... .......... .. 100% 17.1M=0.8s
2018-04-10 10:49:13 (666 KB/s) - ‘ebola-raw.zip’ saved [534670/534670]
%%bash
cd gyak08
bgzip -c ebola-raw/vcf/SNP-2014.vcf > ismertSNPk.vcf.gz
tabix -p vcf ismertSNPk.vcf.gz
Base Quality Score Recalibration (BQSR)¶
%%bash
cd gyak08
export PATH=${PATH}:/home/bioinfo/tools/gatk
gatk BaseRecalibrator \
-R KM034562v1.fa \
-I illesztes01_deduplikalt_grp.bam \
--known-sites ismertSNPk.vcf.gz \
-O rekalibracios.table
Using GATK jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar
Running:
java -Dsamjdk.use_async_io_read_samtools=false -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2 -jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar BaseRecalibrator -R KM034562v1.fa -I illesztes01_deduplikalt_grp.bam --known-sites ismertSNPk.vcf.gz -O rekalibracios.table
10:53:03.990 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar!/com/intel/gkl/native/libgkl_compression.so
10:53:04.112 INFO BaseRecalibrator - ------------------------------------------------------------
10:53:04.112 INFO BaseRecalibrator - The Genome Analysis Toolkit (GATK) v4.0.3.0
10:53:04.112 INFO BaseRecalibrator - For support and documentation go to https://software.broadinstitute.org/gatk/
10:53:04.113 INFO BaseRecalibrator - Executing as sn@sn-OptiPlex-3010 on Linux v4.13.0-38-generic amd64
10:53:04.113 INFO BaseRecalibrator - Java runtime: OpenJDK 64-Bit Server VM v1.8.0_162-8u162-b12-0ubuntu0.16.04.2-b12
10:53:04.113 INFO BaseRecalibrator - Start Date/Time: April 10, 2018 10:53:03 AM CEST
10:53:04.113 INFO BaseRecalibrator - ------------------------------------------------------------
10:53:04.113 INFO BaseRecalibrator - ------------------------------------------------------------
10:53:04.114 INFO BaseRecalibrator - HTSJDK Version: 2.14.3
10:53:04.114 INFO BaseRecalibrator - Picard Version: 2.17.2
10:53:04.114 INFO BaseRecalibrator - HTSJDK Defaults.COMPRESSION_LEVEL : 2
10:53:04.114 INFO BaseRecalibrator - HTSJDK Defaults.USE_ASYNC_IO_READ_FOR_SAMTOOLS : false
10:53:04.114 INFO BaseRecalibrator - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_SAMTOOLS : true
10:53:04.114 INFO BaseRecalibrator - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_TRIBBLE : false
10:53:04.114 INFO BaseRecalibrator - Deflater: IntelDeflater
10:53:04.114 INFO BaseRecalibrator - Inflater: IntelInflater
10:53:04.114 INFO BaseRecalibrator - GCS max retries/reopens: 20
10:53:04.114 INFO BaseRecalibrator - Using google-cloud-java patch 6d11bef1c81f885c26b2b56c8616b7a705171e4f from https://github.com/droazen/google-cloud-java/tree/dr_all_nio_fixes
10:53:04.114 INFO BaseRecalibrator - Initializing engine
10:53:04.452 INFO FeatureManager - Using codec VCFCodec to read file file:///home/sn/gyak08/ismertSNPk.vcf.gz
10:53:04.463 INFO BaseRecalibrator - Shutting down engine
[April 10, 2018 10:53:04 AM CEST] org.broadinstitute.hellbender.tools.walkers.bqsr.BaseRecalibrator done. Elapsed time: 0.01 minutes.
Runtime.totalMemory()=268959744
*******************************************************************
A USER ERROR has occurred: Input files reference and features have incompatible contigs: No overlapping contigs found.
reference contigs = [KM034562v1]
features contigs = [KM034562]
*******************************************************************
Set the system property GATK_STACKTRACE_ON_USER_EXCEPTION (--java-options '-DGATK_STACKTRACE_ON_USER_EXCEPTION=true') to print the stack trace.
## !! TERMINÁL !!
cd gyak08
vim ebola-raw/vcf/SNP-2014.vcf
%%bash
cd gyak08
bgzip -c ebola-raw/vcf/SNP-2014.vcf > ismertSNPk.vcf.gz
tabix -p vcf ismertSNPk.vcf.gz
%%bash
cd gyak08
export PATH=${PATH}:/home/bioinfo/tools/gatk
# Base Quality Score Recalibration (BQSR)
gatk BaseRecalibrator \
-R KM034562v1.fa \
-I illesztes01_deduplikalt_grp.bam \
--known-sites ismertSNPk.vcf.gz \
-O rekalibracios.table
#Apply a linear base quality recalibration model trained with the BaseRecalibrator tool
gatk ApplyBQSR \
-R KM034562v1.fa \
-I illesztes01_deduplikalt_grp.bam \
-bqsr rekalibracios.table \
-O illesztes01_deduplikalt_grp_bqsr.bam
Tool returned:
6302
Using GATK jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar
Running:
java -Dsamjdk.use_async_io_read_samtools=false -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2 -jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar BaseRecalibrator -R KM034562v1.fa -I illesztes01_deduplikalt_grp.bam --known-sites ismertSNPk.vcf.gz -O rekalibracios.table
11:02:38.202 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar!/com/intel/gkl/native/libgkl_compression.so
11:02:38.311 INFO BaseRecalibrator - ------------------------------------------------------------
11:02:38.311 INFO BaseRecalibrator - The Genome Analysis Toolkit (GATK) v4.0.3.0
11:02:38.311 INFO BaseRecalibrator - For support and documentation go to https://software.broadinstitute.org/gatk/
11:02:38.311 INFO BaseRecalibrator - Executing as sn@sn-OptiPlex-3010 on Linux v4.13.0-38-generic amd64
11:02:38.311 INFO BaseRecalibrator - Java runtime: OpenJDK 64-Bit Server VM v1.8.0_162-8u162-b12-0ubuntu0.16.04.2-b12
11:02:38.311 INFO BaseRecalibrator - Start Date/Time: April 10, 2018 11:02:38 AM CEST
11:02:38.312 INFO BaseRecalibrator - ------------------------------------------------------------
11:02:38.312 INFO BaseRecalibrator - ------------------------------------------------------------
11:02:38.312 INFO BaseRecalibrator - HTSJDK Version: 2.14.3
11:02:38.312 INFO BaseRecalibrator - Picard Version: 2.17.2
11:02:38.312 INFO BaseRecalibrator - HTSJDK Defaults.COMPRESSION_LEVEL : 2
11:02:38.312 INFO BaseRecalibrator - HTSJDK Defaults.USE_ASYNC_IO_READ_FOR_SAMTOOLS : false
11:02:38.312 INFO BaseRecalibrator - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_SAMTOOLS : true
11:02:38.312 INFO BaseRecalibrator - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_TRIBBLE : false
11:02:38.312 INFO BaseRecalibrator - Deflater: IntelDeflater
11:02:38.312 INFO BaseRecalibrator - Inflater: IntelInflater
11:02:38.312 INFO BaseRecalibrator - GCS max retries/reopens: 20
11:02:38.312 INFO BaseRecalibrator - Using google-cloud-java patch 6d11bef1c81f885c26b2b56c8616b7a705171e4f from https://github.com/droazen/google-cloud-java/tree/dr_all_nio_fixes
11:02:38.312 INFO BaseRecalibrator - Initializing engine
11:02:38.628 INFO FeatureManager - Using codec VCFCodec to read file file:///home/sn/gyak08/ismertSNPk.vcf.gz
11:02:38.639 INFO BaseRecalibrator - Done initializing engine
11:02:38.642 INFO BaseRecalibrationEngine - The covariates being used here:
11:02:38.642 INFO BaseRecalibrationEngine - ReadGroupCovariate
11:02:38.642 INFO BaseRecalibrationEngine - QualityScoreCovariate
11:02:38.642 INFO BaseRecalibrationEngine - ContextCovariate
11:02:38.642 INFO BaseRecalibrationEngine - CycleCovariate
11:02:38.643 INFO ProgressMeter - Starting traversal
11:02:38.643 INFO ProgressMeter - Current Locus Elapsed Minutes Reads Processed Reads/Minute
11:02:39.090 INFO BaseRecalibrator - No reads filtered by: ((((((MappingQualityNotZeroReadFilter AND MappingQualityAvailableReadFilter) AND MappedReadFilter) AND NotSecondaryAlignmentReadFilter) AND NotDuplicateReadFilter) AND PassesVendorQualityCheckReadFilter) AND WellformedReadFilter)
11:02:39.091 INFO ProgressMeter - KM034562v1:18001 0.0 6302 845906.0
11:02:39.091 INFO ProgressMeter - Traversal complete. Processed 6302 total reads in 0.0 minutes.
11:02:39.163 INFO BaseRecalibrator - Calculating quantized quality scores...
11:02:39.177 INFO BaseRecalibrator - Writing recalibration report...
11:02:39.982 INFO BaseRecalibrator - ...done!
11:02:39.982 INFO BaseRecalibrator - Shutting down engine
[April 10, 2018 11:02:39 AM CEST] org.broadinstitute.hellbender.tools.walkers.bqsr.BaseRecalibrator done. Elapsed time: 0.03 minutes.
Runtime.totalMemory()=276299776
Using GATK jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar
Running:
java -Dsamjdk.use_async_io_read_samtools=false -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2 -jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar ApplyBQSR -R KM034562v1.fa -I illesztes01_deduplikalt_grp.bam -bqsr rekalibracios.table -O illesztes01_deduplikalt_grp_bqsr.bam
11:02:41.832 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar!/com/intel/gkl/native/libgkl_compression.so
11:02:41.931 INFO ApplyBQSR - ------------------------------------------------------------
11:02:41.932 INFO ApplyBQSR - The Genome Analysis Toolkit (GATK) v4.0.3.0
11:02:41.932 INFO ApplyBQSR - For support and documentation go to https://software.broadinstitute.org/gatk/
11:02:41.932 INFO ApplyBQSR - Executing as sn@sn-OptiPlex-3010 on Linux v4.13.0-38-generic amd64
11:02:41.932 INFO ApplyBQSR - Java runtime: OpenJDK 64-Bit Server VM v1.8.0_162-8u162-b12-0ubuntu0.16.04.2-b12
11:02:41.932 INFO ApplyBQSR - Start Date/Time: April 10, 2018 11:02:41 AM CEST
11:02:41.932 INFO ApplyBQSR - ------------------------------------------------------------
11:02:41.932 INFO ApplyBQSR - ------------------------------------------------------------
11:02:41.933 INFO ApplyBQSR - HTSJDK Version: 2.14.3
11:02:41.933 INFO ApplyBQSR - Picard Version: 2.17.2
11:02:41.933 INFO ApplyBQSR - HTSJDK Defaults.COMPRESSION_LEVEL : 2
11:02:41.933 INFO ApplyBQSR - HTSJDK Defaults.USE_ASYNC_IO_READ_FOR_SAMTOOLS : false
11:02:41.933 INFO ApplyBQSR - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_SAMTOOLS : true
11:02:41.933 INFO ApplyBQSR - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_TRIBBLE : false
11:02:41.933 INFO ApplyBQSR - Deflater: IntelDeflater
11:02:41.933 INFO ApplyBQSR - Inflater: IntelInflater
11:02:41.933 INFO ApplyBQSR - GCS max retries/reopens: 20
11:02:41.933 INFO ApplyBQSR - Using google-cloud-java patch 6d11bef1c81f885c26b2b56c8616b7a705171e4f from https://github.com/droazen/google-cloud-java/tree/dr_all_nio_fixes
11:02:41.933 INFO ApplyBQSR - Initializing engine
11:02:42.243 INFO ApplyBQSR - Done initializing engine
11:02:42.254 INFO ProgressMeter - Starting traversal
11:02:42.255 INFO ProgressMeter - Current Locus Elapsed Minutes Reads Processed Reads/Minute
11:02:43.339 INFO ApplyBQSR - No reads filtered by: WellformedReadFilter
11:02:43.340 INFO ProgressMeter - KM034562v1:18001 0.0 6302 348819.2
11:02:43.340 INFO ProgressMeter - Traversal complete. Processed 6302 total reads in 0.0 minutes.
11:02:43.391 INFO ApplyBQSR - Shutting down engine
[April 10, 2018 11:02:43 AM CEST] org.broadinstitute.hellbender.tools.walkers.bqsr.ApplyBQSR done. Elapsed time: 0.03 minutes.
Runtime.totalMemory()=259522560
%%bash
cd gyak08
export PATH=${PATH}:/home/bioinfo/tools/gatk
# Variant calling
gatk HaplotypeCaller \
-R KM034562v1.fa \
-I illesztes01_deduplikalt_grp_bqsr.bam \
-O sznipek_GATK.vcf \
-bamout illesztes01_deduplikalt_grp_bqsr_GATK.bam
Using GATK jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar
Running:
java -Dsamjdk.use_async_io_read_samtools=false -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2 -jar /home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar HaplotypeCaller -R KM034562v1.fa -I illesztes01_deduplikalt_grp_bqsr.bam -O sznipek_GATK.vcf -bamout illesztes01_deduplikalt_grp_bqsr_GATK.bam
11:08:06.998 INFO NativeLibraryLoader - Loading libgkl_compression.so from jar:file:/home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar!/com/intel/gkl/native/libgkl_compression.so
11:08:07.159 INFO HaplotypeCaller - ------------------------------------------------------------
11:08:07.160 INFO HaplotypeCaller - The Genome Analysis Toolkit (GATK) v4.0.3.0
11:08:07.160 INFO HaplotypeCaller - For support and documentation go to https://software.broadinstitute.org/gatk/
11:08:07.160 INFO HaplotypeCaller - Executing as sn@sn-OptiPlex-3010 on Linux v4.13.0-38-generic amd64
11:08:07.160 INFO HaplotypeCaller - Java runtime: OpenJDK 64-Bit Server VM v1.8.0_162-8u162-b12-0ubuntu0.16.04.2-b12
11:08:07.161 INFO HaplotypeCaller - Start Date/Time: April 10, 2018 11:08:06 AM CEST
11:08:07.161 INFO HaplotypeCaller - ------------------------------------------------------------
11:08:07.161 INFO HaplotypeCaller - ------------------------------------------------------------
11:08:07.162 INFO HaplotypeCaller - HTSJDK Version: 2.14.3
11:08:07.162 INFO HaplotypeCaller - Picard Version: 2.17.2
11:08:07.162 INFO HaplotypeCaller - HTSJDK Defaults.COMPRESSION_LEVEL : 2
11:08:07.162 INFO HaplotypeCaller - HTSJDK Defaults.USE_ASYNC_IO_READ_FOR_SAMTOOLS : false
11:08:07.162 INFO HaplotypeCaller - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_SAMTOOLS : true
11:08:07.162 INFO HaplotypeCaller - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_TRIBBLE : false
11:08:07.162 INFO HaplotypeCaller - Deflater: IntelDeflater
11:08:07.162 INFO HaplotypeCaller - Inflater: IntelInflater
11:08:07.162 INFO HaplotypeCaller - GCS max retries/reopens: 20
11:08:07.162 INFO HaplotypeCaller - Using google-cloud-java patch 6d11bef1c81f885c26b2b56c8616b7a705171e4f from https://github.com/droazen/google-cloud-java/tree/dr_all_nio_fixes
11:08:07.162 INFO HaplotypeCaller - Initializing engine
11:08:07.484 INFO HaplotypeCaller - Done initializing engine
11:08:07.487 INFO HaplotypeCallerEngine - Disabling physical phasing, which is supported only for reference-model confidence output
11:08:08.003 INFO NativeLibraryLoader - Loading libgkl_utils.so from jar:file:/home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar!/com/intel/gkl/native/libgkl_utils.so
11:08:08.004 INFO NativeLibraryLoader - Loading libgkl_pairhmm_omp.so from jar:file:/home/bioinfo/tools/gatk/gatk-package-4.0.3.0-local.jar!/com/intel/gkl/native/libgkl_pairhmm_omp.so
11:08:08.046 WARN IntelPairHmm - Flush-to-zero (FTZ) is enabled when running PairHMM
11:08:08.046 INFO IntelPairHmm - Available threads: 4
11:08:08.046 INFO IntelPairHmm - Requested threads: 4
11:08:08.047 INFO PairHMM - Using the OpenMP multi-threaded AVX-accelerated native PairHMM implementation
11:08:08.069 INFO ProgressMeter - Starting traversal
11:08:08.069 INFO ProgressMeter - Current Locus Elapsed Minutes Regions Processed Regions/Minute
11:08:09.300 INFO HaplotypeCaller - No reads filtered by: ((((((((MappingQualityReadFilter AND MappingQualityAvailableReadFilter) AND MappedReadFilter) AND NotSecondaryAlignmentReadFilter) AND NotDuplicateReadFilter) AND PassesVendorQualityCheckReadFilter) AND NonZeroReferenceLengthAlignmentReadFilter) AND GoodCigarReadFilter) AND WellformedReadFilter)
11:08:09.300 INFO ProgressMeter - KM034562v1:17105 0.0 78 3801.8
11:08:09.300 INFO ProgressMeter - Traversal complete. Processed 78 total regions in 0.0 minutes.
11:08:09.302 INFO VectorLoglessPairHMM - Time spent in setup for JNI call : 0.0011534140000000002
11:08:09.302 INFO PairHMM - Total compute time in PairHMM computeLogLikelihoods() : 0.026732288000000003
11:08:09.302 INFO SmithWatermanAligner - Total compute time in java Smith-Waterman : 0.04 sec
11:08:09.380 INFO HaplotypeCaller - Shutting down engine
[April 10, 2018 11:08:09 AM CEST] org.broadinstitute.hellbender.tools.walkers.haplotypecaller.HaplotypeCaller done. Elapsed time: 0.04 minutes.
Runtime.totalMemory()=397934592
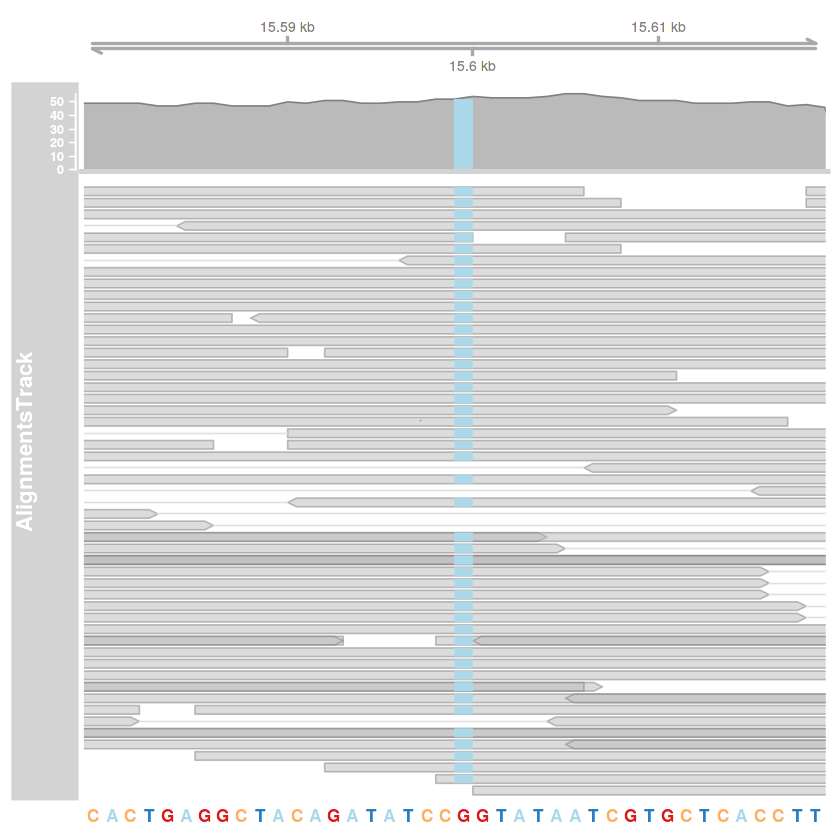
# R
library(Gviz)
library(seqinr)
library(Biostrings)
options(ucscChromosomeNames=FALSE)
setwd('gyak08')
# a 15599. pozíció ábrázolása
poz = 15599
kezdet = poz - 20
veg = poz + 20
bam.fajlom = 'illesztes01_deduplikalt_grp_bqsr.bam'
illesztes.track = AlignmentsTrack(bam.fajlom, start=kezdet, end=veg)
tengely.track = GenomeAxisTrack()
referencia = read.fasta('KM034562v1.fa', as.string=TRUE, seqonly=TRUE)
referencia.szekvencia = referencia[[1]]
referencia.szekvencia = DNAStringSet(referencia.szekvencia)
names(referencia.szekvencia) = 'KM034562v1'
szekvencia.track = SequenceTrack(referencia.szekvencia)
plotTracks(
list(tengely.track, illesztes.track, szekvencia.track),
type=c('coverage', 'pileup'),
chromosome='KM034562v1',
from=kezdet,
to=veg
)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, cbind, colMeans, colnames,
colSums, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, lengths, Map, mapply, match,
mget, order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rowMeans, rownames, rowSums, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which, which.max, which.min
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:base’:
expand.grid
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: grid
Loading required package: XVector
Attaching package: 'Biostrings'
The following object is masked from 'package:seqinr':
translate
The following object is masked from 'package:base':
strsplit
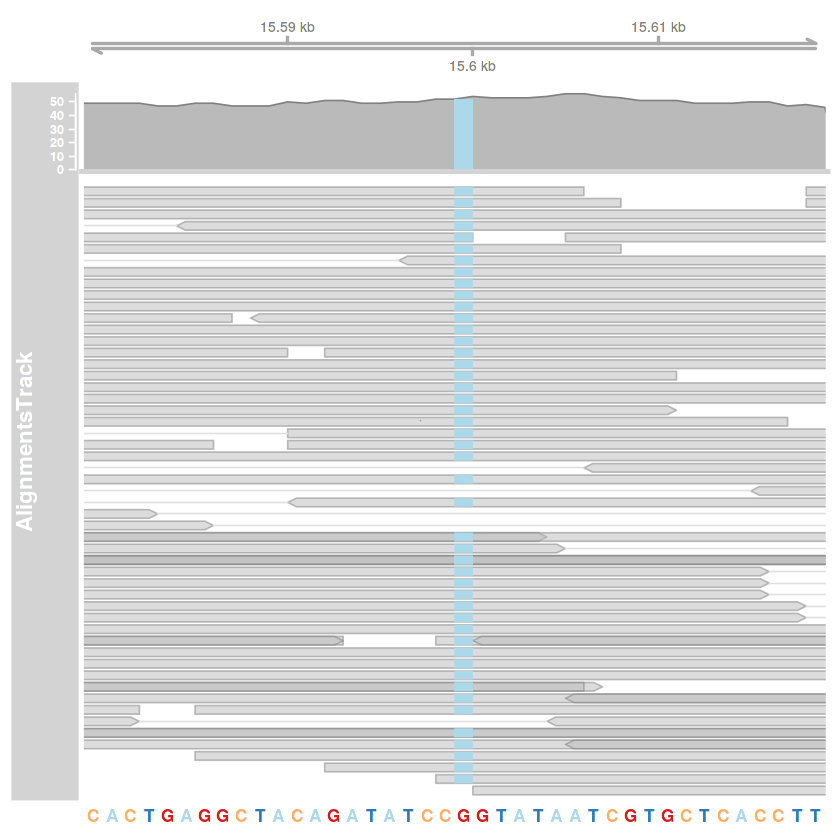
bam.fajlom = 'illesztes01_deduplikalt_grp_bqsr_GATK.bam'
illesztes.track = AlignmentsTrack(bam.fajlom, start=kezdet, end=veg)
plotTracks(
list(tengely.track, illesztes.track, szekvencia.track),
type=c('coverage', 'pileup'),
chromosome='KM034562v1',
from=kezdet,
to=veg
)